LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_3.5wLII_08492_1
Total number of 3D structures: 67
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
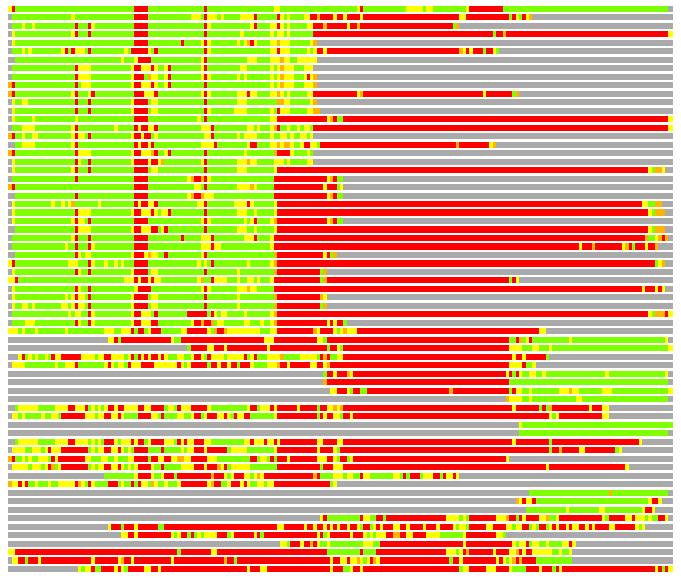
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3c3w_A |
211 |
200 |
182 |
0.94 |
16.48 |
89.042 |
17.515 |
T P |
| 1a2o_A |
347 |
200 |
94 |
1.81 |
11.70 |
42.637 |
4.934 |
T P |
| 2gwr_A |
225 |
200 |
87 |
1.69 |
8.05 |
41.047 |
4.849 |
T P |
| 1w25_A |
454 |
200 |
87 |
1.45 |
10.34 |
40.945 |
5.599 |
T P |
| 1qmp_A |
125 |
200 |
85 |
1.58 |
4.71 |
40.068 |
5.062 |
T P |
| 1p2f_A |
217 |
200 |
88 |
1.78 |
6.82 |
39.549 |
4.674 |
T P |
| 3eul_B |
124 |
200 |
86 |
1.68 |
11.63 |
39.535 |
4.832 |
T P |
| 1ab5_A |
125 |
200 |
85 |
1.68 |
17.65 |
39.439 |
4.765 |
T P |
| 1vlz_A |
128 |
200 |
85 |
1.79 |
17.65 |
39.423 |
4.500 |
T P |
| 1d4z_A |
128 |
200 |
86 |
1.87 |
17.44 |
39.200 |
4.372 |
T P |
| 1ys7_B |
226 |
200 |
88 |
1.90 |
9.09 |
39.188 |
4.407 |
T P |
| 2jba_A |
125 |
200 |
85 |
1.80 |
12.94 |
38.773 |
4.470 |
T P |
| 1b00_A |
122 |
200 |
85 |
1.75 |
12.94 |
38.750 |
4.582 |
T P |
| 3cz5_C |
142 |
200 |
80 |
1.28 |
16.25 |
38.667 |
5.816 |
T P |
| 3f6c_A |
129 |
200 |
85 |
1.79 |
14.12 |
38.323 |
4.507 |
T P |
| 1ny5_B |
385 |
200 |
84 |
1.77 |
10.71 |
37.734 |
4.484 |
T P |
| 1s8n_A |
190 |
200 |
85 |
1.96 |
14.12 |
37.578 |
4.122 |
T P |
| 1ymu_A |
125 |
200 |
81 |
1.69 |
18.52 |
37.539 |
4.516 |
T P |
| 1yio_A |
198 |
200 |
82 |
1.69 |
8.54 |
37.428 |
4.582 |
T P |
| 1zes_A |
121 |
200 |
78 |
1.50 |
12.82 |
37.342 |
4.867 |
T P |
| 1u0s_Y |
118 |
200 |
75 |
1.45 |
16.00 |
36.930 |
4.829 |
T P |
| 1dz3_A |
123 |
200 |
77 |
1.53 |
6.49 |
36.914 |
4.728 |
T P |
| 1tmy_A |
118 |
200 |
75 |
1.53 |
16.00 |
36.671 |
4.599 |
T P |
| 3b2n_A |
120 |
200 |
80 |
1.83 |
8.75 |
36.611 |
4.137 |
T P |
| 2j8e_A |
128 |
200 |
79 |
1.83 |
16.46 |
36.508 |
4.091 |
T P |
| 2a9o_A |
117 |
200 |
76 |
1.48 |
13.16 |
36.406 |
4.821 |
T P |
| 1mih_A |
128 |
200 |
77 |
1.77 |
16.88 |
36.325 |
4.116 |
T P |
| 2a9r_A |
117 |
200 |
76 |
1.62 |
11.84 |
36.305 |
4.406 |
T P |
| 2oqr_A |
226 |
200 |
79 |
1.80 |
8.86 |
35.986 |
4.163 |
T P |
| 2chy_A |
128 |
200 |
76 |
1.67 |
18.42 |
35.921 |
4.295 |
T P |
| 3bre_B |
328 |
200 |
77 |
1.69 |
9.09 |
35.478 |
4.295 |
T P |
| 2iyn_B |
124 |
200 |
75 |
1.51 |
13.33 |
35.355 |
4.662 |
T P |
| 1a04_A |
205 |
200 |
78 |
1.86 |
11.54 |
35.283 |
3.988 |
T P |
| 1mvo_A |
121 |
200 |
75 |
1.62 |
13.33 |
35.237 |
4.354 |
T P |
| 2jb9_B |
122 |
200 |
75 |
1.63 |
13.33 |
34.954 |
4.338 |
T P |
| 2jba_B |
121 |
200 |
75 |
1.72 |
13.33 |
34.511 |
4.128 |
T P |
| 2qsj_B |
122 |
200 |
75 |
1.87 |
12.00 |
33.841 |
3.811 |
T P |
| 1dc7_A |
124 |
200 |
73 |
1.98 |
8.22 |
31.509 |
3.518 |
T P |
| 3g13_A |
152 |
200 |
78 |
2.31 |
8.97 |
28.681 |
3.231 |
T P |
| 1p4w_A |
87 |
200 |
58 |
1.80 |
18.97 |
26.761 |
3.059 |
T P |
| 1s7o_C |
108 |
200 |
57 |
1.64 |
22.81 |
25.573 |
3.274 |
T P |
| 2r0q_C |
193 |
200 |
78 |
2.61 |
5.13 |
24.634 |
2.882 |
T P |
| 3bvp_A |
136 |
200 |
71 |
2.51 |
7.04 |
24.413 |
2.715 |
T P |
| 1x3u_A |
79 |
200 |
51 |
1.67 |
21.57 |
23.842 |
2.880 |
T P |
| 1je8_E |
67 |
200 |
49 |
1.18 |
20.41 |
23.811 |
3.837 |
T P |
| 2hwv_A |
101 |
200 |
55 |
1.97 |
12.73 |
23.137 |
2.660 |
T P |
| 2rnj_A |
67 |
200 |
49 |
1.53 |
22.45 |
23.090 |
3.007 |
T P |
| 2gm5_A |
122 |
200 |
71 |
2.63 |
5.63 |
23.078 |
2.597 |
T P |
| 1gdt_A |
183 |
200 |
67 |
2.33 |
10.45 |
22.994 |
2.760 |
T P |
| 3c57_A |
51 |
200 |
46 |
0.76 |
23.91 |
22.686 |
5.367 |
T P |
| 1zlj_A |
69 |
200 |
45 |
0.52 |
24.44 |
22.500 |
7.277 |
T P |
| 2rsl_B |
120 |
200 |
67 |
2.64 |
5.97 |
21.976 |
2.445 |
T P |
| 1zr4_A |
183 |
200 |
63 |
2.40 |
11.11 |
21.671 |
2.519 |
T P |
| 1ght_A |
105 |
200 |
64 |
2.40 |
4.69 |
21.244 |
2.560 |
T P |
| 2gm4_A |
179 |
200 |
62 |
2.48 |
8.06 |
21.136 |
2.399 |
T P |
| 2zb9_A |
188 |
200 |
59 |
2.30 |
6.78 |
21.118 |
2.460 |
T P |
| 1dc8_A |
123 |
200 |
59 |
2.45 |
10.17 |
20.226 |
2.318 |
T P |
| 2jpc_A |
61 |
200 |
42 |
1.29 |
26.19 |
20.035 |
3.020 |
T P |
| 1res_A |
43 |
200 |
40 |
1.67 |
25.00 |
18.535 |
2.266 |
T P |
| 1jko_C |
46 |
200 |
37 |
1.43 |
18.92 |
17.327 |
2.424 |
T P |
| 2jn6_A |
97 |
200 |
47 |
2.32 |
6.38 |
17.079 |
1.944 |
T P |
| 3cwo_X |
236 |
200 |
51 |
2.62 |
7.84 |
17.052 |
1.873 |
T P |
| 2rae_A |
193 |
200 |
48 |
2.63 |
4.17 |
16.542 |
1.759 |
T P |
| 1umq_A |
60 |
200 |
42 |
2.12 |
7.14 |
15.742 |
1.892 |
T P |
| 1fse_B |
70 |
200 |
42 |
2.37 |
7.14 |
15.715 |
1.700 |
T P |
| 3clo_A |
258 |
200 |
41 |
2.66 |
12.20 |
14.205 |
1.484 |
T P |
| 2elh_A |
87 |
200 |
37 |
2.64 |
8.11 |
12.998 |
1.350 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]