LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_300.5wLII_11238_61
Total number of 3D structures: 46
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
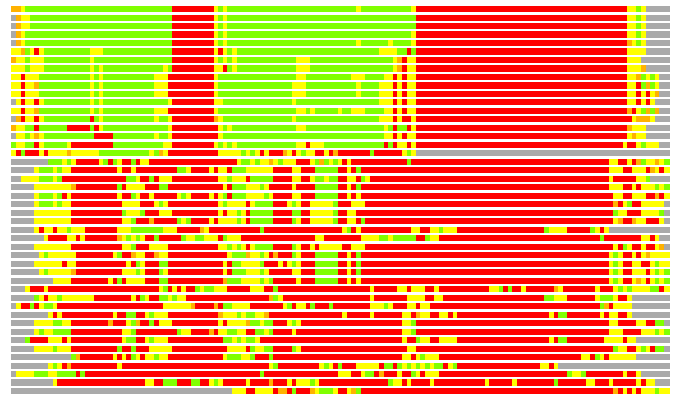
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3b8w_A |
358 |
143 |
83 |
1.36 |
9.64 |
56.143 |
5.667 |
T P |
| 2rjg_A |
358 |
143 |
82 |
1.19 |
9.76 |
55.680 |
6.373 |
T P |
| 3b8t_A |
358 |
143 |
82 |
1.20 |
10.98 |
55.634 |
6.313 |
T P |
| 3b8v_A |
358 |
143 |
82 |
1.24 |
9.76 |
55.579 |
6.121 |
T P |
| 3b8u_A |
358 |
143 |
82 |
1.26 |
9.76 |
55.573 |
6.014 |
T P |
| 1vfs_A |
382 |
143 |
80 |
1.63 |
6.25 |
53.365 |
4.617 |
T P |
| 1rcq_A |
356 |
143 |
81 |
1.82 |
6.17 |
52.395 |
4.219 |
T P |
| 2odo_A |
354 |
143 |
81 |
1.88 |
8.64 |
51.853 |
4.099 |
T P |
| 2sfp_A |
378 |
143 |
82 |
1.99 |
6.10 |
51.528 |
3.925 |
T P |
| 1epv_A |
381 |
143 |
81 |
1.99 |
6.17 |
51.232 |
3.881 |
T P |
| 1xql_A |
381 |
143 |
80 |
1.95 |
6.25 |
51.081 |
3.908 |
T P |
| 2vd9_A |
386 |
143 |
78 |
1.84 |
5.13 |
50.996 |
4.020 |
T P |
| 1bd0_A |
381 |
143 |
81 |
2.07 |
6.17 |
50.181 |
3.729 |
T P |
| 2vd8_A |
386 |
143 |
77 |
1.97 |
6.49 |
50.021 |
3.727 |
T P |
| 1xfc_A |
366 |
143 |
73 |
1.59 |
4.11 |
49.096 |
4.314 |
T P |
| 2dy3_D |
344 |
143 |
75 |
1.86 |
8.00 |
48.799 |
3.826 |
T P |
| 3co8_A |
360 |
143 |
71 |
1.96 |
11.27 |
43.767 |
3.451 |
T P |
| 1foh_C |
656 |
143 |
51 |
2.54 |
7.84 |
24.415 |
1.930 |
T P |
| 1bgj_A |
391 |
143 |
45 |
2.55 |
2.22 |
20.910 |
1.700 |
T P |
| 1bf3_A |
391 |
143 |
43 |
2.65 |
9.30 |
20.845 |
1.561 |
T P |
| 2vou_A |
393 |
143 |
43 |
2.55 |
6.98 |
20.597 |
1.620 |
T P |
| 1dob_A |
394 |
143 |
43 |
2.67 |
6.98 |
20.546 |
1.551 |
T P |
| 1pbe_A |
391 |
143 |
42 |
2.60 |
9.52 |
20.503 |
1.553 |
T P |
| 1pxc_A |
394 |
143 |
46 |
2.59 |
8.70 |
20.319 |
1.713 |
T P |
| 1cc4_A |
391 |
143 |
44 |
2.65 |
6.82 |
20.286 |
1.602 |
T P |
| 1pbd_A |
391 |
143 |
44 |
2.75 |
4.55 |
20.243 |
1.543 |
T P |
| 3c96_A |
381 |
143 |
44 |
2.63 |
4.55 |
20.172 |
1.611 |
T P |
| 2rgj_A |
376 |
143 |
44 |
2.70 |
6.82 |
19.767 |
1.574 |
T P |
| 1doc_A |
394 |
143 |
43 |
2.63 |
4.65 |
19.617 |
1.577 |
T P |
| 1ykj_A |
394 |
143 |
45 |
2.83 |
2.22 |
19.602 |
1.537 |
T P |
| 1cj2_A |
391 |
143 |
41 |
2.59 |
4.88 |
19.575 |
1.522 |
T P |
| 1bkw_A |
391 |
143 |
43 |
2.67 |
2.33 |
19.569 |
1.554 |
T P |
| 1k0i_A |
394 |
143 |
41 |
2.67 |
2.44 |
19.347 |
1.479 |
T P |
| 1pn0_C |
656 |
143 |
43 |
2.83 |
9.30 |
18.960 |
1.466 |
T P |
| 2qa2_A |
489 |
143 |
42 |
2.69 |
2.38 |
18.599 |
1.503 |
T P |
| 2dkh_A |
614 |
143 |
41 |
2.78 |
7.32 |
18.579 |
1.426 |
T P |
| 1cc6_A |
391 |
143 |
38 |
2.84 |
5.26 |
18.471 |
1.292 |
T P |
| 1cj3_A |
392 |
143 |
39 |
2.65 |
7.69 |
18.451 |
1.417 |
T P |
| 1cj4_A |
392 |
143 |
38 |
2.58 |
5.26 |
18.097 |
1.417 |
T P |
| 1bgn_A |
391 |
143 |
38 |
2.75 |
10.53 |
17.849 |
1.333 |
T P |
| 1pxb_A |
394 |
143 |
36 |
2.53 |
5.56 |
17.665 |
1.366 |
T P |
| 1pbf_A |
391 |
143 |
36 |
2.69 |
8.33 |
16.366 |
1.290 |
T P |
| 2qa1_A |
488 |
143 |
33 |
2.59 |
3.03 |
16.343 |
1.228 |
T P |
| 2hai_A |
562 |
143 |
34 |
2.59 |
5.88 |
16.201 |
1.264 |
T P |
| 2r0c_A |
509 |
143 |
33 |
2.83 |
3.03 |
15.099 |
1.127 |
T P |
| 1pxa_A |
394 |
143 |
28 |
2.88 |
3.57 |
13.527 |
0.938 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]