LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_303.5wLII_11238_73
Total number of 3D structures: 50
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
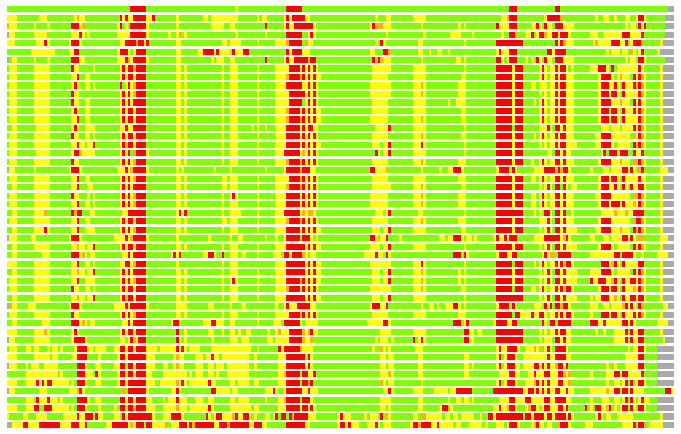
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1krh_A |
337 |
248 |
229 |
0.35 |
23.14 |
92.363 |
50.881 |
T P |
| 2r6h_C |
289 |
248 |
221 |
1.73 |
19.91 |
81.123 |
12.065 |
T P |
| 1fdr_A |
244 |
248 |
214 |
1.84 |
16.82 |
79.153 |
11.056 |
T P |
| 1gvh_A |
396 |
248 |
216 |
1.83 |
17.59 |
78.053 |
11.174 |
T P |
| 2rc5_A |
306 |
248 |
208 |
1.78 |
21.15 |
75.667 |
11.038 |
T P |
| 1qfj_A |
226 |
248 |
208 |
1.80 |
22.60 |
75.448 |
10.974 |
T P |
| 3fpk_A |
247 |
248 |
212 |
1.79 |
16.98 |
73.836 |
11.237 |
T P |
| 1gjr_A |
295 |
248 |
211 |
1.91 |
17.06 |
73.787 |
10.501 |
T P |
| 1go2_A |
295 |
248 |
210 |
1.90 |
17.62 |
73.583 |
10.500 |
T P |
| 1e63_A |
295 |
248 |
210 |
1.91 |
17.14 |
73.485 |
10.452 |
T P |
| 1ogj_A |
295 |
248 |
210 |
1.91 |
17.14 |
73.269 |
10.455 |
T P |
| 1qh0_A |
295 |
248 |
210 |
1.91 |
17.62 |
73.212 |
10.423 |
T P |
| 1h85_A |
295 |
248 |
208 |
1.88 |
17.79 |
72.980 |
10.500 |
T P |
| 1gr1_A |
295 |
248 |
209 |
1.90 |
17.70 |
72.964 |
10.429 |
T P |
| 2b5o_A |
292 |
248 |
213 |
1.86 |
19.72 |
72.879 |
10.855 |
T P |
| 1bqe_A |
295 |
248 |
210 |
1.92 |
18.10 |
72.692 |
10.372 |
T P |
| 1e64_A |
295 |
248 |
207 |
1.90 |
18.36 |
72.532 |
10.361 |
T P |
| 1jb9_A |
307 |
248 |
205 |
1.75 |
18.54 |
71.872 |
11.053 |
T P |
| 1que_A |
303 |
248 |
212 |
1.96 |
16.98 |
71.702 |
10.308 |
T P |
| 1a8p_A |
257 |
248 |
207 |
1.81 |
20.29 |
71.701 |
10.852 |
T P |
| 1b2r_A |
295 |
248 |
210 |
1.93 |
17.14 |
71.512 |
10.347 |
T P |
| 1quf_A |
296 |
248 |
208 |
1.90 |
17.31 |
71.178 |
10.381 |
T P |
| 1qgz_A |
295 |
248 |
210 |
1.95 |
17.62 |
71.080 |
10.255 |
T P |
| 1e62_A |
295 |
248 |
208 |
1.92 |
17.31 |
70.897 |
10.275 |
T P |
| 1gaw_B |
305 |
248 |
206 |
1.85 |
21.36 |
70.838 |
10.554 |
T P |
| 1ogi_A |
295 |
248 |
210 |
1.92 |
18.57 |
70.694 |
10.376 |
T P |
| 1h42_A |
295 |
248 |
208 |
1.92 |
18.75 |
70.570 |
10.291 |
T P |
| 2qdx_A |
257 |
248 |
207 |
1.85 |
20.29 |
70.491 |
10.589 |
T P |
| 1qgy_A |
295 |
248 |
207 |
1.92 |
18.36 |
70.234 |
10.225 |
T P |
| 1ep3_B |
261 |
248 |
203 |
1.94 |
19.21 |
70.181 |
9.972 |
T P |
| 1ewy_A |
303 |
248 |
206 |
1.90 |
17.48 |
70.022 |
10.284 |
T P |
| 2bmw_A |
295 |
248 |
207 |
1.94 |
19.32 |
69.879 |
10.145 |
T P |
| 1w34_A |
295 |
248 |
205 |
1.91 |
18.05 |
69.862 |
10.194 |
T P |
| 1bjk_A |
295 |
248 |
208 |
1.92 |
17.31 |
69.713 |
10.297 |
T P |
| 2bsa_A |
295 |
248 |
204 |
1.93 |
18.63 |
69.180 |
10.057 |
T P |
| 2bgi_A |
257 |
248 |
209 |
1.94 |
17.70 |
68.925 |
10.240 |
T P |
| 1w35_A |
295 |
248 |
207 |
1.98 |
17.39 |
68.132 |
9.931 |
T P |
| 1ep1_B |
261 |
248 |
200 |
1.91 |
18.00 |
67.778 |
9.954 |
T P |
| 1ddg_A |
374 |
248 |
210 |
1.92 |
18.57 |
67.538 |
10.421 |
T P |
| 1tll_A |
630 |
248 |
209 |
1.87 |
16.27 |
67.187 |
10.610 |
T P |
| 2eix_A |
243 |
248 |
209 |
2.05 |
17.70 |
65.130 |
9.721 |
T P |
| 1qx4_A |
264 |
248 |
209 |
2.00 |
20.10 |
64.754 |
9.955 |
T P |
| 1umk_A |
271 |
248 |
209 |
2.00 |
18.18 |
64.669 |
9.931 |
T P |
| 2cnd_A |
260 |
248 |
210 |
2.10 |
18.57 |
62.499 |
9.529 |
T P |
| 1i7p_A |
272 |
248 |
203 |
2.08 |
20.69 |
61.208 |
9.329 |
T P |
| 2pia_A |
321 |
248 |
195 |
2.05 |
21.03 |
60.312 |
9.056 |
T P |
| 1cne_A |
260 |
248 |
206 |
2.06 |
18.93 |
60.134 |
9.551 |
T P |
| 1ndh_A |
270 |
248 |
196 |
2.15 |
18.37 |
55.672 |
8.703 |
T P |
| 1tvc_A |
250 |
248 |
182 |
2.28 |
22.53 |
49.405 |
7.650 |
T P |
| 1cqx_A |
403 |
248 |
165 |
2.48 |
26.67 |
46.652 |
6.394 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]