LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_309.5wLII_11238_93
Total number of 3D structures: 52
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
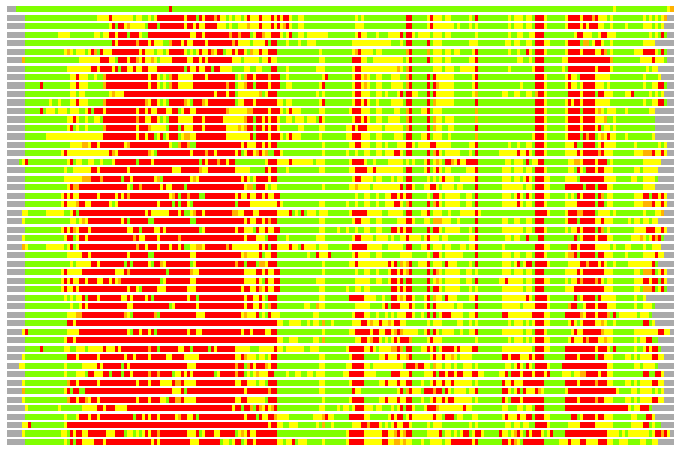
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2p11_A |
219 |
222 |
218 |
0.44 |
44.04 |
97.839 |
40.427 |
T P |
| 2w4m_A |
250 |
222 |
175 |
1.99 |
14.86 |
57.724 |
8.373 |
T P |
| 2gfh_A |
246 |
222 |
174 |
2.09 |
14.94 |
56.260 |
7.958 |
T P |
| 2hoq_A |
237 |
222 |
159 |
2.06 |
23.90 |
56.184 |
7.367 |
T P |
| 3ed5_A |
232 |
222 |
167 |
2.09 |
19.16 |
55.845 |
7.618 |
T P |
| 1x42_A |
230 |
222 |
165 |
2.23 |
16.97 |
54.661 |
7.088 |
T P |
| 2om6_A |
233 |
222 |
165 |
2.15 |
16.36 |
53.959 |
7.341 |
T P |
| 3ddh_A |
229 |
222 |
162 |
2.17 |
19.75 |
52.269 |
7.150 |
T P |
| 1aq6_A |
245 |
222 |
155 |
2.02 |
16.77 |
51.698 |
7.298 |
T P |
| 1qq7_A |
244 |
222 |
155 |
2.08 |
16.77 |
51.500 |
7.110 |
T P |
| 2pib_A |
216 |
222 |
155 |
2.03 |
11.61 |
51.412 |
7.282 |
T P |
| 1qq5_A |
245 |
222 |
151 |
2.03 |
16.56 |
51.195 |
7.104 |
T P |
| 2no5_B |
225 |
222 |
150 |
2.17 |
13.33 |
50.842 |
6.611 |
T P |
| 1qh9_A |
220 |
222 |
158 |
2.14 |
13.92 |
50.751 |
7.046 |
T P |
| 1zrn_A |
220 |
222 |
156 |
2.11 |
14.10 |
50.687 |
7.058 |
T P |
| 2no4_B |
226 |
222 |
155 |
2.24 |
13.55 |
50.383 |
6.610 |
T P |
| 2nyv_A |
217 |
222 |
152 |
2.08 |
18.42 |
49.202 |
6.962 |
T P |
| 2ioh_A |
256 |
222 |
152 |
2.34 |
14.47 |
46.978 |
6.220 |
T P |
| 1qyi_A |
380 |
222 |
152 |
2.10 |
13.16 |
46.672 |
6.899 |
T P |
| 2hsz_A |
225 |
222 |
149 |
2.15 |
18.79 |
46.534 |
6.611 |
T P |
| 2ah5_A |
210 |
222 |
145 |
2.18 |
17.24 |
46.510 |
6.353 |
T P |
| 3e58_A |
211 |
222 |
137 |
2.17 |
13.14 |
46.505 |
6.043 |
T P |
| 1fez_A |
256 |
222 |
139 |
2.12 |
15.83 |
46.004 |
6.272 |
T P |
| 1rdf_A |
263 |
222 |
146 |
2.26 |
12.33 |
45.844 |
6.190 |
T P |
| 2w11_A |
206 |
222 |
144 |
2.26 |
13.89 |
45.692 |
6.095 |
T P |
| 2yy6_A |
208 |
222 |
143 |
2.02 |
18.18 |
45.531 |
6.743 |
T P |
| 2iof_A |
256 |
222 |
141 |
2.09 |
14.89 |
45.001 |
6.429 |
T P |
| 2hcf_A |
225 |
222 |
139 |
2.12 |
16.55 |
44.922 |
6.274 |
T P |
| 2w43_A |
201 |
222 |
149 |
2.45 |
14.09 |
44.914 |
5.836 |
T P |
| 2fdr_A |
222 |
222 |
145 |
2.19 |
13.10 |
44.880 |
6.325 |
T P |
| 2hi0_A |
240 |
222 |
143 |
2.17 |
11.89 |
44.858 |
6.296 |
T P |
| 1rql_A |
257 |
222 |
143 |
2.24 |
13.99 |
44.194 |
6.105 |
T P |
| 2iof_K |
255 |
222 |
141 |
2.21 |
14.89 |
43.947 |
6.097 |
T P |
| 1swv_A |
257 |
222 |
138 |
2.11 |
13.77 |
43.906 |
6.244 |
T P |
| 3d6j_A |
210 |
222 |
133 |
2.06 |
21.80 |
42.958 |
6.170 |
T P |
| 2hdo_A |
207 |
222 |
137 |
2.14 |
11.68 |
42.941 |
6.103 |
T P |
| 1o08_A |
221 |
222 |
133 |
2.31 |
12.03 |
42.777 |
5.529 |
T P |
| 1wvi_A |
253 |
222 |
126 |
2.07 |
13.49 |
42.366 |
5.817 |
T P |
| 3dv9_A |
243 |
222 |
136 |
2.18 |
14.71 |
42.289 |
5.961 |
T P |
| 1ydf_A |
255 |
222 |
123 |
2.00 |
13.01 |
42.017 |
5.865 |
T P |
| 1lvh_A |
220 |
222 |
139 |
2.31 |
10.79 |
41.818 |
5.760 |
T P |
| 1j97_A |
210 |
222 |
136 |
2.23 |
16.91 |
41.791 |
5.837 |
T P |
| 2go7_C |
205 |
222 |
135 |
2.26 |
15.56 |
41.678 |
5.716 |
T P |
| 1te2_A |
218 |
222 |
136 |
2.35 |
11.76 |
41.628 |
5.544 |
T P |
| 1f5s_A |
210 |
222 |
134 |
2.26 |
17.16 |
41.409 |
5.675 |
T P |
| 2zg6_B |
203 |
222 |
122 |
2.04 |
13.93 |
41.071 |
5.709 |
T P |
| 1l7m_B |
210 |
222 |
133 |
2.21 |
16.54 |
40.927 |
5.751 |
T P |
| 2fi1_A |
187 |
222 |
125 |
2.16 |
12.80 |
40.591 |
5.540 |
T P |
| 2qlt_A |
251 |
222 |
129 |
2.27 |
13.18 |
39.771 |
5.448 |
T P |
| 1yv9_A |
257 |
222 |
124 |
2.07 |
15.32 |
39.769 |
5.723 |
T P |
| 2b8e_C |
254 |
222 |
136 |
2.42 |
14.71 |
39.063 |
5.407 |
T P |
| 2iye_A |
249 |
222 |
129 |
2.34 |
13.95 |
37.441 |
5.297 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]