LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_313.5wLII_11238_106
Total number of 3D structures: 48
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
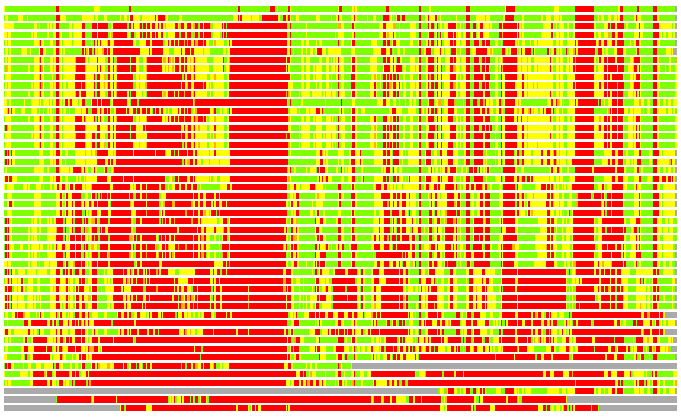
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2ivd_A |
449 |
463 |
418 |
0.95 |
16.27 |
87.949 |
39.876 |
T P |
| 1sez_A |
465 |
463 |
394 |
1.88 |
13.96 |
66.297 |
19.947 |
T P |
| 2iid_A |
483 |
463 |
306 |
2.19 |
12.42 |
44.419 |
13.357 |
T P |
| 1reo_A |
484 |
463 |
307 |
2.17 |
11.73 |
44.166 |
13.545 |
T P |
| 2bxr_A |
445 |
463 |
307 |
2.26 |
12.05 |
43.302 |
12.995 |
T P |
| 2b9w_A |
423 |
463 |
310 |
2.22 |
10.65 |
42.780 |
13.383 |
T P |
| 2z5y_A |
513 |
463 |
307 |
2.41 |
11.07 |
42.765 |
12.222 |
T P |
| 2z5x_A |
513 |
463 |
307 |
2.44 |
11.40 |
42.463 |
12.068 |
T P |
| 2c72_A |
499 |
463 |
293 |
2.37 |
13.65 |
40.977 |
11.874 |
T P |
| 1o5w_C |
512 |
463 |
291 |
2.40 |
11.68 |
40.829 |
11.661 |
T P |
| 2c75_A |
499 |
463 |
301 |
2.45 |
13.29 |
40.730 |
11.792 |
T P |
| 2jae_A |
478 |
463 |
276 |
2.11 |
13.41 |
40.699 |
12.469 |
T P |
| 2yr5_A |
684 |
463 |
304 |
2.39 |
11.51 |
40.660 |
12.201 |
T P |
| 1s3e_A |
499 |
463 |
299 |
2.41 |
13.71 |
40.253 |
11.891 |
T P |
| 2c73_A |
499 |
463 |
294 |
2.43 |
12.93 |
40.166 |
11.623 |
T P |
| 2bk5_A |
499 |
463 |
295 |
2.42 |
13.22 |
40.139 |
11.727 |
T P |
| 2c76_A |
499 |
463 |
294 |
2.41 |
12.93 |
39.830 |
11.727 |
T P |
| 2vvm_A |
481 |
463 |
292 |
2.41 |
13.70 |
38.897 |
11.638 |
T P |
| 2vvl_G |
478 |
463 |
286 |
2.42 |
15.38 |
38.347 |
11.347 |
T P |
| 1yvv_A |
328 |
463 |
261 |
2.05 |
13.03 |
37.942 |
12.118 |
T P |
| 2vvl_A |
478 |
463 |
289 |
2.42 |
14.19 |
37.763 |
11.462 |
T P |
| 1b37_B |
462 |
463 |
281 |
2.42 |
13.88 |
37.428 |
11.162 |
T P |
| 3bnm_B |
499 |
463 |
259 |
2.34 |
10.81 |
37.166 |
10.597 |
T P |
| 1yy5_A |
498 |
463 |
263 |
2.43 |
12.17 |
36.932 |
10.407 |
T P |
| 1rsg_B |
491 |
463 |
256 |
2.33 |
9.77 |
36.189 |
10.547 |
T P |
| 2v1d_A |
666 |
463 |
257 |
2.29 |
13.62 |
36.188 |
10.765 |
T P |
| 2iw5_A |
666 |
463 |
256 |
2.26 |
13.67 |
36.168 |
10.848 |
T P |
| 2h94_A |
647 |
463 |
253 |
2.28 |
13.04 |
36.129 |
10.617 |
T P |
| 2z3y_A |
643 |
463 |
257 |
2.44 |
13.23 |
35.893 |
10.130 |
T P |
| 2dw4_A |
634 |
463 |
243 |
2.31 |
14.40 |
34.999 |
10.090 |
T P |
| 2hko_A |
647 |
463 |
248 |
2.33 |
14.52 |
34.739 |
10.214 |
T P |
| 1i8t_A |
367 |
463 |
242 |
2.51 |
7.02 |
31.514 |
9.289 |
T P |
| 1v0j_A |
388 |
463 |
231 |
2.42 |
12.12 |
30.781 |
9.173 |
T P |
| 1usj_A |
376 |
463 |
233 |
2.53 |
8.58 |
30.365 |
8.864 |
T P |
| 1wam_A |
377 |
463 |
228 |
2.50 |
8.77 |
29.971 |
8.758 |
T P |
| 2bi7_A |
383 |
463 |
222 |
2.46 |
9.01 |
29.902 |
8.688 |
T P |
| 2bcg_G |
442 |
463 |
215 |
2.55 |
10.70 |
29.430 |
8.100 |
T P |
| 2oln_A |
385 |
463 |
199 |
2.36 |
13.07 |
28.537 |
8.083 |
T P |
| 3cpi_H |
439 |
463 |
202 |
2.55 |
10.89 |
27.385 |
7.632 |
T P |
| 1pj5_A |
827 |
463 |
190 |
2.32 |
15.26 |
26.658 |
7.842 |
T P |
| 2q6u_A |
383 |
463 |
203 |
2.55 |
11.82 |
26.439 |
7.649 |
T P |
| 1knr_A |
529 |
463 |
157 |
2.12 |
12.10 |
23.228 |
7.084 |
T P |
| 2e1m_A |
356 |
463 |
139 |
2.28 |
20.14 |
20.574 |
5.834 |
T P |
| 1w4x_A |
533 |
463 |
140 |
2.36 |
10.00 |
20.168 |
5.698 |
T P |
| 2gjc_A |
301 |
463 |
132 |
2.21 |
15.15 |
19.172 |
5.712 |
T P |
| 2e1m_C |
151 |
463 |
124 |
2.34 |
8.06 |
18.031 |
5.077 |
T P |
| 1zrh_A |
263 |
463 |
51 |
2.53 |
5.88 |
7.248 |
1.938 |
T P |
| 2pmi_C |
285 |
463 |
44 |
2.68 |
6.82 |
6.330 |
1.584 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]