LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_314.5wLII_11238_109
Total number of 3D structures: 89
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
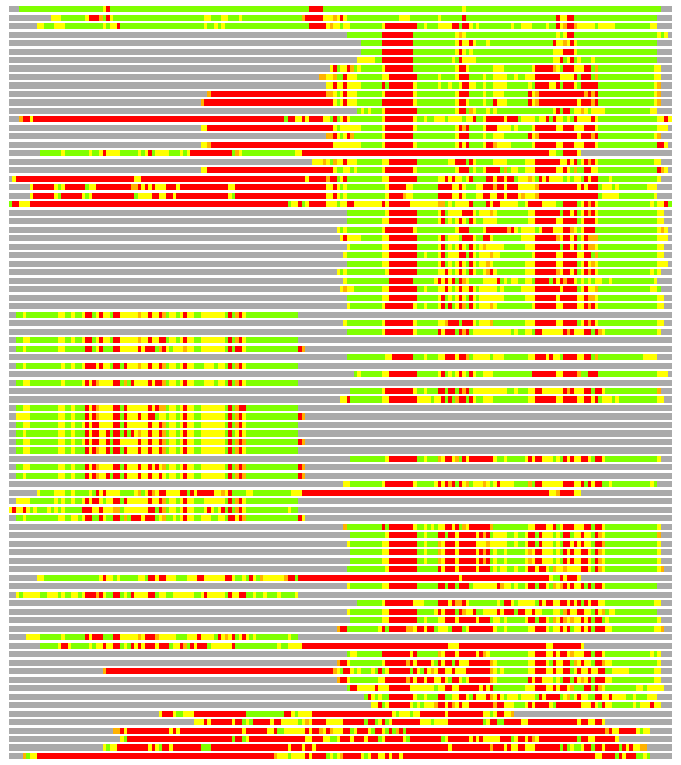
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1m70_A |
190 |
190 |
179 |
0.37 |
29.05 |
94.047 |
38.230 |
T P |
| 1h1o_A |
172 |
190 |
160 |
1.46 |
32.50 |
80.150 |
10.277 |
T P |
| 1fcd_C |
174 |
190 |
156 |
1.90 |
19.87 |
68.666 |
7.811 |
T P |
| 1cno_G |
87 |
190 |
81 |
1.15 |
35.80 |
41.600 |
6.460 |
T P |
| 1c53_A |
79 |
190 |
72 |
1.34 |
33.33 |
37.013 |
5.004 |
T P |
| 1dvh_A |
79 |
190 |
72 |
1.32 |
29.17 |
36.990 |
5.059 |
T P |
| 2dvh_A |
79 |
190 |
74 |
1.65 |
28.38 |
36.027 |
4.239 |
T P |
| 1e2r_B |
543 |
190 |
73 |
1.93 |
21.92 |
34.371 |
3.595 |
T P |
| 1hzu_A |
521 |
190 |
75 |
2.11 |
21.33 |
33.771 |
3.389 |
T P |
| 1hzv_A |
514 |
190 |
71 |
2.09 |
21.13 |
32.027 |
3.238 |
T P |
| 1gq1_A |
559 |
190 |
69 |
2.09 |
23.19 |
31.516 |
3.146 |
T P |
| 1qks_A |
559 |
190 |
66 |
1.98 |
25.76 |
31.389 |
3.177 |
T P |
| 1e08_E |
78 |
190 |
72 |
1.94 |
27.78 |
31.031 |
3.530 |
T P |
| 1kv9_A |
664 |
190 |
82 |
2.39 |
18.29 |
30.022 |
3.299 |
T P |
| 1mg2_D |
147 |
190 |
73 |
1.96 |
20.55 |
29.943 |
3.541 |
T P |
| 1hj5_A |
559 |
190 |
67 |
1.99 |
23.88 |
29.797 |
3.207 |
T P |
| 2gc4_D |
147 |
190 |
73 |
1.94 |
20.55 |
29.776 |
3.573 |
T P |
| 1c52_A |
131 |
190 |
66 |
1.93 |
22.73 |
29.524 |
3.254 |
T P |
| 1nir_B |
539 |
190 |
76 |
2.18 |
25.00 |
29.418 |
3.328 |
T P |
| 2d0w_A |
168 |
190 |
73 |
2.06 |
16.44 |
29.173 |
3.383 |
T P |
| 1kb0_A |
669 |
190 |
75 |
2.22 |
16.00 |
28.602 |
3.239 |
T P |
| 2c1d_A |
264 |
190 |
78 |
2.44 |
15.38 |
28.455 |
3.072 |
T P |
| 1h32_A |
261 |
190 |
77 |
2.34 |
15.58 |
28.404 |
3.154 |
T P |
| 1yiq_A |
684 |
190 |
84 |
2.54 |
17.86 |
28.346 |
3.183 |
T P |
| 1c6r_A |
88 |
190 |
67 |
1.90 |
23.88 |
28.177 |
3.354 |
T P |
| 1f1f_A |
88 |
190 |
67 |
1.86 |
22.39 |
27.989 |
3.411 |
T P |
| 1umm_A |
149 |
190 |
69 |
1.98 |
15.94 |
27.988 |
3.310 |
T P |
| 2ce0_A |
99 |
190 |
69 |
2.12 |
21.74 |
27.819 |
3.101 |
T P |
| 1gdv_A |
85 |
190 |
69 |
1.98 |
17.39 |
27.627 |
3.321 |
T P |
| 1ctj_A |
89 |
190 |
68 |
2.02 |
25.00 |
27.619 |
3.211 |
T P |
| 2zbo_A |
86 |
190 |
70 |
1.97 |
18.57 |
27.594 |
3.385 |
T P |
| 1ls9_A |
91 |
190 |
70 |
2.01 |
21.43 |
27.413 |
3.311 |
T P |
| 1w2l_A |
97 |
190 |
70 |
1.99 |
22.86 |
27.232 |
3.356 |
T P |
| 2dge_A |
102 |
190 |
69 |
2.12 |
17.39 |
26.927 |
3.107 |
T P |
| 2v08_A |
84 |
190 |
68 |
2.05 |
22.06 |
26.902 |
3.169 |
T P |
| 1cyj_A |
90 |
190 |
67 |
1.96 |
26.87 |
26.557 |
3.254 |
T P |
| 1chh_A |
108 |
190 |
71 |
2.29 |
16.90 |
26.425 |
2.976 |
T P |
| 1kib_A |
89 |
190 |
66 |
1.95 |
19.70 |
26.341 |
3.225 |
T P |
| 1wej_F |
104 |
190 |
69 |
2.15 |
24.64 |
26.306 |
3.062 |
T P |
| 1irv_A |
108 |
190 |
70 |
2.27 |
17.14 |
26.289 |
2.949 |
T P |
| 2bcn_B |
108 |
190 |
69 |
2.16 |
15.94 |
26.215 |
3.056 |
T P |
| 2c1d_B |
137 |
190 |
68 |
2.14 |
20.59 |
26.191 |
3.032 |
T P |
| 1s6v_B |
108 |
190 |
69 |
2.21 |
17.39 |
26.042 |
2.981 |
T P |
| 2v07_A |
98 |
190 |
65 |
1.90 |
23.08 |
26.039 |
3.252 |
T P |
| 3cxh_W |
112 |
190 |
70 |
2.32 |
15.71 |
25.789 |
2.892 |
T P |
| 1yeb_A |
108 |
190 |
67 |
2.13 |
14.93 |
25.615 |
3.007 |
T P |
| 1c6s_A |
87 |
190 |
66 |
2.02 |
22.73 |
25.606 |
3.120 |
T P |
| 1crh_A |
108 |
190 |
70 |
2.35 |
15.71 |
25.564 |
2.859 |
T P |
| 1crg_A |
108 |
190 |
71 |
2.39 |
14.08 |
25.555 |
2.846 |
T P |
| 1ctz_A |
108 |
190 |
70 |
2.32 |
15.71 |
25.533 |
2.889 |
T P |
| 1csw_A |
108 |
190 |
68 |
2.27 |
16.18 |
25.527 |
2.869 |
T P |
| 1chj_A |
108 |
190 |
70 |
2.36 |
15.71 |
25.491 |
2.845 |
T P |
| 1yea_A |
112 |
190 |
69 |
2.31 |
13.04 |
25.481 |
2.862 |
T P |
| 1raq_A |
108 |
190 |
62 |
2.11 |
14.52 |
25.466 |
2.804 |
T P |
| 1irw_A |
108 |
190 |
71 |
2.42 |
14.08 |
25.455 |
2.818 |
T P |
| 1cig_A |
108 |
190 |
70 |
2.35 |
14.29 |
25.453 |
2.855 |
T P |
| 1cyc_A |
103 |
190 |
68 |
2.23 |
27.94 |
25.299 |
2.917 |
T P |
| 2fwl_A |
129 |
190 |
67 |
2.44 |
23.88 |
25.271 |
2.636 |
T P |
| 1ytc_A |
112 |
190 |
70 |
2.35 |
15.71 |
25.189 |
2.856 |
T P |
| 2ycc_A |
108 |
190 |
69 |
2.40 |
18.84 |
25.152 |
2.755 |
T P |
| 1csv_A |
108 |
190 |
67 |
2.13 |
14.93 |
25.068 |
3.002 |
T P |
| 1i54_A |
103 |
190 |
63 |
2.10 |
30.16 |
24.885 |
2.868 |
T P |
| 2b11_B |
108 |
190 |
63 |
2.15 |
12.70 |
24.807 |
2.802 |
T P |
| 3cx5_W |
108 |
190 |
65 |
2.19 |
15.38 |
24.803 |
2.843 |
T P |
| 1ycc_A |
108 |
190 |
64 |
2.19 |
14.06 |
24.792 |
2.790 |
T P |
| 2jqr_A |
108 |
190 |
64 |
2.19 |
14.06 |
24.792 |
2.790 |
T P |
| 2b4z_A |
104 |
190 |
64 |
2.12 |
26.56 |
24.781 |
2.878 |
T P |
| 1dt1_A |
129 |
190 |
69 |
2.38 |
15.94 |
24.532 |
2.777 |
T P |
| 2b0z_B |
108 |
190 |
63 |
2.10 |
17.46 |
24.383 |
2.859 |
T P |
| 1yfc_A |
108 |
190 |
68 |
2.28 |
17.65 |
24.369 |
2.858 |
T P |
| 1wve_C |
75 |
190 |
62 |
2.20 |
24.19 |
24.246 |
2.691 |
T P |
| 2jti_B |
103 |
190 |
64 |
2.27 |
12.50 |
24.073 |
2.697 |
T P |
| 2b12_B |
108 |
190 |
61 |
2.09 |
18.03 |
23.979 |
2.779 |
T P |
| 1cri_A |
108 |
190 |
62 |
2.32 |
8.06 |
23.772 |
2.561 |
T P |
| 1j3s_A |
104 |
190 |
65 |
2.31 |
13.85 |
23.725 |
2.697 |
T P |
| 1foc_A |
128 |
190 |
62 |
2.38 |
25.81 |
23.648 |
2.502 |
T P |
| 1ccr_A |
111 |
190 |
62 |
2.16 |
20.97 |
23.601 |
2.742 |
T P |
| 1rap_A |
108 |
190 |
61 |
2.31 |
8.20 |
23.524 |
2.533 |
T P |
| 1fhb_A |
108 |
190 |
64 |
2.49 |
6.25 |
23.100 |
2.468 |
T P |
| 1csu_A |
108 |
190 |
61 |
2.35 |
8.20 |
23.077 |
2.490 |
T P |
| 2aiu_A |
104 |
190 |
63 |
2.52 |
17.46 |
22.672 |
2.402 |
T P |
| 1a56_A |
81 |
190 |
60 |
2.52 |
13.33 |
20.574 |
2.294 |
T P |
| 1lms_A |
95 |
190 |
48 |
2.62 |
8.33 |
17.006 |
1.763 |
T P |
| 2occ_B |
227 |
190 |
40 |
2.39 |
7.50 |
16.004 |
1.605 |
T P |
| 1v54_B |
227 |
190 |
46 |
2.85 |
8.70 |
15.806 |
1.562 |
T P |
| 2gsm_B |
256 |
190 |
39 |
2.93 |
7.69 |
13.745 |
1.286 |
T P |
| 3ehb_B |
252 |
190 |
36 |
2.90 |
5.56 |
12.770 |
1.199 |
T P |
| 1qle_B |
252 |
190 |
36 |
2.71 |
8.33 |
12.685 |
1.282 |
T P |
| 1m56_B |
260 |
190 |
34 |
2.67 |
2.94 |
11.772 |
1.227 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]