LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_317.5wLII_11247_7
Total number of 3D structures: 49
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
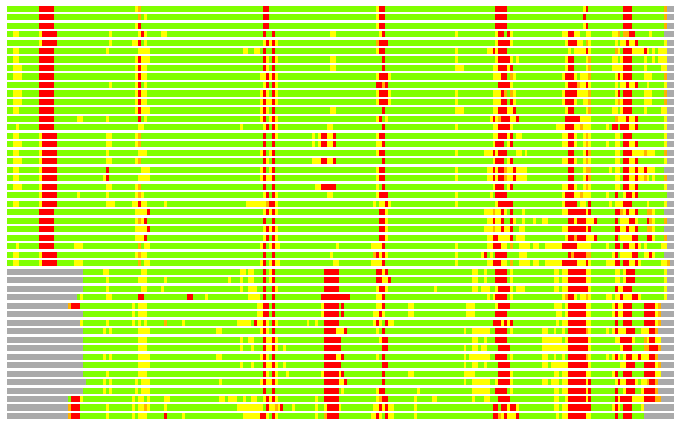
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1yau_A |
222 |
229 |
210 |
0.63 |
22.86 |
91.071 |
28.599 |
T P |
| 1yar_A |
221 |
229 |
210 |
0.67 |
22.86 |
90.968 |
27.382 |
T P |
| 1ya7_A |
227 |
229 |
209 |
0.60 |
22.97 |
90.910 |
29.759 |
T P |
| 1iru_C |
250 |
229 |
210 |
1.39 |
16.67 |
87.357 |
14.081 |
T P |
| 1j2q_A |
237 |
229 |
207 |
1.27 |
19.32 |
86.963 |
15.076 |
T P |
| 1iru_F |
238 |
229 |
209 |
1.41 |
11.48 |
86.484 |
13.842 |
T P |
| 3bdm_B |
244 |
229 |
209 |
1.32 |
19.62 |
86.438 |
14.708 |
T P |
| 1g0u_B |
235 |
229 |
209 |
1.36 |
19.62 |
86.323 |
14.342 |
T P |
| 1ryp_D |
241 |
229 |
207 |
1.38 |
17.39 |
86.313 |
13.963 |
T P |
| 1j2p_A |
243 |
229 |
206 |
1.26 |
19.90 |
86.245 |
15.107 |
T P |
| 1g0u_C |
238 |
229 |
207 |
1.41 |
17.87 |
86.195 |
13.694 |
T P |
| 3bdm_C |
241 |
229 |
207 |
1.42 |
17.39 |
86.034 |
13.616 |
T P |
| 1ryp_C |
244 |
229 |
209 |
1.38 |
19.62 |
85.917 |
14.132 |
T P |
| 1iru_A |
244 |
229 |
206 |
1.40 |
9.71 |
85.477 |
13.773 |
T P |
| 1ryp_B |
250 |
229 |
206 |
1.39 |
17.48 |
85.450 |
13.818 |
T P |
| 1iru_E |
234 |
229 |
206 |
1.40 |
16.99 |
85.182 |
13.726 |
T P |
| 1ryp_E |
242 |
229 |
207 |
1.42 |
16.43 |
85.044 |
13.603 |
T P |
| 1g0u_E |
230 |
229 |
208 |
1.50 |
14.90 |
85.003 |
12.967 |
T P |
| 3bdm_D |
242 |
229 |
207 |
1.44 |
16.43 |
84.816 |
13.410 |
T P |
| 1ryp_F |
233 |
229 |
206 |
1.52 |
15.05 |
84.781 |
12.680 |
T P |
| 3bdm_E |
233 |
229 |
207 |
1.54 |
14.98 |
84.753 |
12.593 |
T P |
| 1g0u_D |
230 |
229 |
205 |
1.40 |
17.07 |
84.442 |
13.631 |
T P |
| 1iru_B |
233 |
229 |
206 |
1.37 |
14.56 |
84.440 |
14.052 |
T P |
| 1iru_D |
243 |
229 |
205 |
1.49 |
14.63 |
84.322 |
12.863 |
T P |
| 1g0u_F |
242 |
229 |
203 |
1.49 |
13.79 |
84.278 |
12.754 |
T P |
| 1z7q_G |
243 |
229 |
204 |
1.45 |
14.22 |
84.165 |
13.174 |
T P |
| 1ryp_G |
244 |
229 |
203 |
1.51 |
13.79 |
83.885 |
12.638 |
T P |
| 3bdm_F |
244 |
229 |
203 |
1.47 |
14.29 |
83.760 |
12.914 |
T P |
| 1g0u_G |
240 |
229 |
206 |
1.61 |
12.14 |
83.632 |
12.069 |
T P |
| 1iru_G |
245 |
229 |
203 |
1.52 |
15.27 |
83.628 |
12.557 |
T P |
| 1ryp_A |
243 |
229 |
206 |
1.64 |
11.65 |
83.556 |
11.866 |
T P |
| 1pma_B |
203 |
229 |
178 |
1.38 |
15.17 |
73.608 |
12.059 |
T P |
| 1yar_H |
203 |
229 |
178 |
1.40 |
15.17 |
73.439 |
11.867 |
T P |
| 1j2q_H |
202 |
229 |
177 |
1.50 |
19.21 |
72.058 |
11.032 |
T P |
| 2z5c_C |
189 |
229 |
177 |
1.50 |
15.25 |
71.553 |
11.067 |
T P |
| 1ryp_H |
205 |
229 |
173 |
1.52 |
8.09 |
71.317 |
10.688 |
T P |
| 1g65_N |
196 |
229 |
171 |
1.47 |
8.19 |
70.639 |
10.893 |
T P |
| 1g0u_L |
222 |
229 |
171 |
1.61 |
15.79 |
70.515 |
10.027 |
T P |
| 1g0u_K |
212 |
229 |
174 |
1.63 |
14.37 |
70.495 |
10.073 |
T P |
| 1iru_I |
220 |
229 |
172 |
1.45 |
13.37 |
70.390 |
11.132 |
T P |
| 1ryp_I |
222 |
229 |
172 |
1.48 |
12.79 |
70.325 |
10.898 |
T P |
| 1ryp_L |
212 |
229 |
174 |
1.64 |
14.37 |
70.323 |
10.017 |
T P |
| 3bdm_H |
222 |
229 |
171 |
1.43 |
12.87 |
70.064 |
11.176 |
T P |
| 1iru_H |
202 |
229 |
172 |
1.46 |
10.47 |
70.047 |
10.998 |
T P |
| 1g65_K |
211 |
229 |
171 |
1.63 |
14.62 |
69.490 |
9.894 |
T P |
| 1iru_L |
201 |
229 |
170 |
1.50 |
14.12 |
69.457 |
10.608 |
T P |
| 1iru_J |
204 |
229 |
172 |
1.68 |
11.05 |
69.392 |
9.659 |
T P |
| 1iru_M |
213 |
229 |
169 |
1.67 |
12.43 |
69.056 |
9.544 |
T P |
| 1ryp_M |
222 |
229 |
171 |
1.68 |
14.62 |
67.977 |
9.615 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]