LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_32.5wLII_10799_15
Total number of 3D structures: 54
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
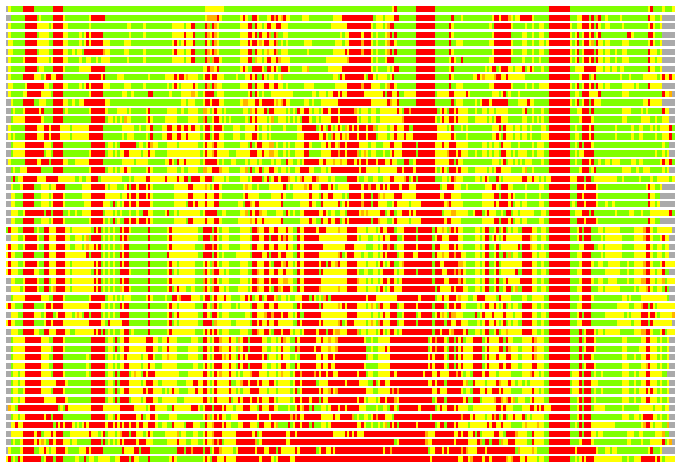
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1sbp_A |
309 |
282 |
253 |
0.91 |
23.32 |
87.746 |
24.962 |
T P |
| 2h5y_B |
233 |
282 |
204 |
1.89 |
13.73 |
60.898 |
10.244 |
T P |
| 3cg1_A |
293 |
282 |
213 |
1.88 |
9.86 |
58.407 |
10.739 |
T P |
| 3cfz_A |
289 |
282 |
207 |
1.92 |
12.08 |
56.427 |
10.249 |
T P |
| 2ons_A |
314 |
282 |
210 |
2.00 |
12.86 |
56.241 |
10.010 |
T P |
| 3cfx_A |
293 |
282 |
207 |
2.01 |
11.59 |
55.331 |
9.825 |
T P |
| 3cg3_A |
315 |
282 |
213 |
2.01 |
7.98 |
54.950 |
10.093 |
T P |
| 1amf_A |
231 |
282 |
203 |
1.88 |
10.34 |
54.599 |
10.242 |
T P |
| 2qry_C |
318 |
282 |
215 |
2.10 |
10.23 |
53.821 |
9.761 |
T P |
| 3cij_A |
291 |
282 |
206 |
2.04 |
13.59 |
53.609 |
9.624 |
T P |
| 3c9h_B |
339 |
282 |
217 |
2.12 |
11.52 |
53.162 |
9.779 |
T P |
| 1atg_A |
231 |
282 |
194 |
2.14 |
10.82 |
49.331 |
8.668 |
T P |
| 1d9y_A |
309 |
282 |
208 |
2.23 |
11.54 |
49.188 |
8.918 |
T P |
| 1mrp_A |
309 |
282 |
210 |
2.29 |
10.00 |
48.249 |
8.788 |
T P |
| 1poy_1 |
323 |
282 |
203 |
2.24 |
9.36 |
47.956 |
8.687 |
T P |
| 1pot_A |
322 |
282 |
205 |
2.28 |
9.27 |
47.948 |
8.624 |
T P |
| 1q35_A |
317 |
282 |
202 |
2.24 |
11.39 |
47.714 |
8.616 |
T P |
| 1y4t_A |
317 |
282 |
207 |
2.38 |
9.18 |
47.618 |
8.345 |
T P |
| 1a99_A |
341 |
282 |
204 |
2.32 |
7.35 |
46.855 |
8.416 |
T P |
| 1xvx_A |
311 |
282 |
201 |
2.24 |
11.94 |
46.754 |
8.584 |
T P |
| 1eu8_A |
407 |
282 |
205 |
2.49 |
9.27 |
44.947 |
7.917 |
T P |
| 3fir_A |
231 |
282 |
190 |
2.29 |
14.74 |
44.655 |
7.956 |
T P |
| 2hxw_B |
230 |
282 |
192 |
2.34 |
14.06 |
44.398 |
7.859 |
T P |
| 3fjm_A |
231 |
282 |
190 |
2.30 |
11.05 |
44.318 |
7.911 |
T P |
| 2v84_A |
319 |
282 |
198 |
2.40 |
8.59 |
44.270 |
7.922 |
T P |
| 3fj7_A |
231 |
282 |
190 |
2.30 |
10.53 |
44.009 |
7.930 |
T P |
| 3c4m_A |
469 |
282 |
196 |
2.47 |
8.67 |
43.781 |
7.641 |
T P |
| 3ehu_A |
441 |
282 |
190 |
2.44 |
7.37 |
43.258 |
7.492 |
T P |
| 3ehs_A |
457 |
282 |
196 |
2.50 |
8.67 |
42.916 |
7.536 |
T P |
| 3eht_A |
452 |
282 |
189 |
2.43 |
8.47 |
42.766 |
7.469 |
T P |
| 1r6z_Z |
499 |
282 |
192 |
2.45 |
7.81 |
42.693 |
7.526 |
T P |
| 1mdp_1 |
363 |
282 |
190 |
2.45 |
7.89 |
42.654 |
7.463 |
T P |
| 1hsj_A |
487 |
282 |
191 |
2.46 |
8.38 |
42.646 |
7.456 |
T P |
| 3dm0_A |
675 |
282 |
191 |
2.45 |
7.85 |
42.474 |
7.501 |
T P |
| 1xvy_A |
307 |
282 |
185 |
2.37 |
10.81 |
42.201 |
7.477 |
T P |
| 3d4g_E |
472 |
282 |
190 |
2.45 |
6.84 |
42.110 |
7.461 |
T P |
| 1mg1_A |
450 |
282 |
189 |
2.44 |
8.47 |
41.905 |
7.444 |
T P |
| 2vgq_A |
461 |
282 |
187 |
2.42 |
6.42 |
41.777 |
7.426 |
T P |
| 3f5f_A |
656 |
282 |
186 |
2.43 |
9.14 |
41.683 |
7.346 |
T P |
| 2o68_A |
307 |
282 |
173 |
2.50 |
12.14 |
40.184 |
6.664 |
T P |
| 1qw0_A |
308 |
282 |
171 |
2.52 |
12.28 |
40.029 |
6.524 |
T P |
| 2o6a_A |
307 |
282 |
170 |
2.44 |
12.35 |
40.017 |
6.704 |
T P |
| 1qvs_A |
308 |
282 |
171 |
2.46 |
11.70 |
39.916 |
6.681 |
T P |
| 1xc1_A |
309 |
282 |
178 |
2.60 |
10.67 |
39.197 |
6.604 |
T P |
| 1nnf_A |
308 |
282 |
171 |
2.59 |
10.53 |
38.827 |
6.362 |
T P |
| 1o7t_A |
309 |
282 |
176 |
2.56 |
10.23 |
38.424 |
6.607 |
T P |
| 2o69_A |
308 |
282 |
164 |
2.53 |
11.59 |
38.155 |
6.236 |
T P |
| 2pt1_A |
317 |
282 |
177 |
2.60 |
10.73 |
37.834 |
6.561 |
T P |
| 1y9u_A |
314 |
282 |
152 |
2.45 |
9.87 |
35.712 |
5.960 |
T P |
| 2voz_B |
315 |
282 |
163 |
2.64 |
14.11 |
35.621 |
5.941 |
T P |
| 2heu_A |
391 |
282 |
158 |
2.50 |
6.96 |
34.963 |
6.071 |
T P |
| 2hq0_A |
388 |
282 |
139 |
2.40 |
9.35 |
33.117 |
5.552 |
T P |
| 1twy_A |
249 |
282 |
133 |
2.39 |
3.01 |
31.764 |
5.346 |
T P |
| 2nvu_B |
789 |
282 |
129 |
2.51 |
7.75 |
29.343 |
4.946 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]