LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_320.5wLII_11276_9
Total number of 3D structures: 86
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
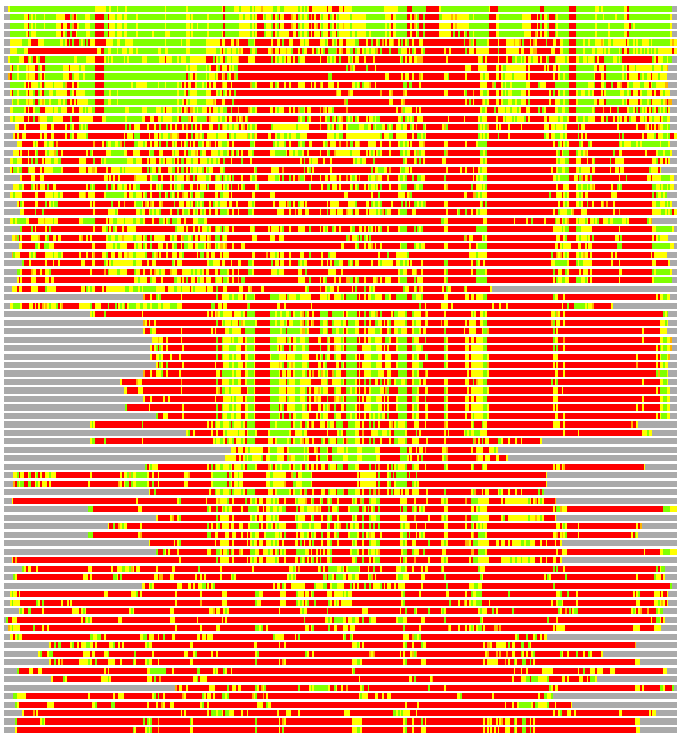
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1dp4_C |
429 |
377 |
343 |
1.70 |
14.29 |
83.760 |
19.067 |
T P |
| 1jdp_A |
396 |
377 |
313 |
2.02 |
11.82 |
60.540 |
14.750 |
T P |
| 1yk0_A |
394 |
377 |
311 |
2.00 |
11.90 |
60.043 |
14.803 |
T P |
| 1yk1_A |
394 |
377 |
309 |
1.97 |
12.30 |
59.637 |
14.907 |
T P |
| 1ewk_B |
449 |
377 |
242 |
2.32 |
10.33 |
42.390 |
9.981 |
T P |
| 2e4z_A |
434 |
377 |
236 |
2.44 |
11.44 |
40.258 |
9.288 |
T P |
| 2e4u_B |
514 |
377 |
217 |
2.39 |
9.68 |
39.696 |
8.719 |
T P |
| 2lbp_A |
346 |
377 |
202 |
2.16 |
14.36 |
38.665 |
8.957 |
T P |
| 2liv_A |
344 |
377 |
198 |
2.12 |
13.64 |
38.598 |
8.929 |
T P |
| 1pea_A |
368 |
377 |
213 |
2.39 |
13.15 |
37.963 |
8.541 |
T P |
| 1z15_A |
344 |
377 |
195 |
2.25 |
12.82 |
37.581 |
8.306 |
T P |
| 1usg_A |
345 |
377 |
193 |
2.25 |
13.47 |
37.035 |
8.203 |
T P |
| 1qnl_A |
368 |
377 |
201 |
2.43 |
12.94 |
35.991 |
7.943 |
T P |
| 3eaf_A |
384 |
377 |
215 |
2.48 |
9.30 |
35.174 |
8.342 |
T P |
| 2h4a_A |
317 |
377 |
160 |
2.66 |
6.25 |
28.817 |
5.796 |
T P |
| 3ckm_A |
318 |
377 |
178 |
2.68 |
6.74 |
28.787 |
6.394 |
T P |
| 1tjy_A |
316 |
377 |
154 |
2.58 |
7.14 |
26.360 |
5.747 |
T P |
| 3e3m_A |
270 |
377 |
160 |
2.64 |
5.00 |
26.048 |
5.845 |
T P |
| 2qvc_A |
302 |
377 |
141 |
2.55 |
7.80 |
23.671 |
5.312 |
T P |
| 3c6q_B |
305 |
377 |
143 |
2.75 |
6.99 |
22.956 |
5.020 |
T P |
| 2h3h_A |
313 |
377 |
142 |
2.68 |
11.97 |
22.925 |
5.105 |
T P |
| 2fn9_A |
280 |
377 |
136 |
2.52 |
6.62 |
22.500 |
5.184 |
T P |
| 1dbp_A |
271 |
377 |
124 |
2.81 |
4.84 |
21.871 |
4.263 |
T P |
| 3clk_B |
262 |
377 |
128 |
2.74 |
7.81 |
21.270 |
4.503 |
T P |
| 2fqx_A |
316 |
377 |
129 |
2.72 |
9.30 |
20.881 |
4.581 |
T P |
| 3eif_A |
936 |
377 |
119 |
2.44 |
12.61 |
20.810 |
4.694 |
T P |
| 1drk_A |
271 |
377 |
123 |
2.58 |
6.50 |
20.771 |
4.587 |
T P |
| 2dri_A |
271 |
377 |
123 |
2.76 |
4.88 |
20.770 |
4.298 |
T P |
| 2fn8_A |
292 |
377 |
120 |
2.60 |
5.83 |
20.724 |
4.448 |
T P |
| 2gx6_A |
271 |
377 |
128 |
2.92 |
7.03 |
20.563 |
4.241 |
T P |
| 1ba2_A |
271 |
377 |
124 |
2.68 |
2.42 |
20.527 |
4.466 |
T P |
| 1drj_A |
271 |
377 |
120 |
2.60 |
3.33 |
20.494 |
4.439 |
T P |
| 2ioy_A |
274 |
377 |
115 |
2.58 |
9.57 |
19.601 |
4.289 |
T P |
| 2qh8_A |
296 |
377 |
117 |
2.65 |
8.55 |
19.375 |
4.260 |
T P |
| 1g7w_A |
396 |
377 |
113 |
2.48 |
10.62 |
19.061 |
4.375 |
T P |
| 1rrv_A |
401 |
377 |
105 |
2.53 |
7.62 |
18.910 |
3.996 |
T P |
| 2q7w_A |
378 |
377 |
107 |
2.40 |
11.21 |
18.811 |
4.283 |
T P |
| 1asf_A |
396 |
377 |
110 |
2.52 |
10.91 |
18.726 |
4.196 |
T P |
| 2aat_A |
396 |
377 |
104 |
2.40 |
11.54 |
18.710 |
4.161 |
T P |
| 1ix8_A |
396 |
377 |
111 |
2.46 |
11.71 |
18.534 |
4.340 |
T P |
| 1g4v_A |
396 |
377 |
106 |
2.35 |
10.38 |
18.225 |
4.325 |
T P |
| 1ix6_A |
396 |
377 |
112 |
2.51 |
7.14 |
18.220 |
4.295 |
T P |
| 5eaa_A |
396 |
377 |
108 |
2.46 |
9.26 |
18.182 |
4.213 |
T P |
| 1spa_A |
396 |
377 |
110 |
2.55 |
8.18 |
18.165 |
4.151 |
T P |
| 2d64_A |
396 |
377 |
105 |
2.45 |
10.48 |
18.005 |
4.120 |
T P |
| 1ars_A |
396 |
377 |
110 |
2.51 |
9.09 |
18.003 |
4.214 |
T P |
| 1ahx_A |
396 |
377 |
106 |
2.50 |
11.32 |
17.985 |
4.070 |
T P |
| 1bqd_A |
395 |
377 |
104 |
2.37 |
10.58 |
17.817 |
4.212 |
T P |
| 1g7x_A |
396 |
377 |
102 |
2.35 |
8.82 |
17.479 |
4.170 |
T P |
| 2d66_A |
396 |
377 |
97 |
2.38 |
9.28 |
17.399 |
3.913 |
T P |
| 1itz_A |
666 |
377 |
98 |
2.42 |
11.22 |
17.155 |
3.882 |
T P |
| 1g4x_A |
396 |
377 |
96 |
2.47 |
6.25 |
17.075 |
3.729 |
T P |
| 3dm9_B |
305 |
377 |
94 |
2.36 |
10.64 |
16.991 |
3.824 |
T P |
| 3e70_C |
307 |
377 |
94 |
2.39 |
9.57 |
16.848 |
3.771 |
T P |
| 2d5y_A |
396 |
377 |
97 |
2.52 |
10.31 |
16.783 |
3.703 |
T P |
| 1wdw_B |
385 |
377 |
98 |
2.52 |
5.10 |
16.565 |
3.744 |
T P |
| 1v8z_B |
387 |
377 |
97 |
2.49 |
4.12 |
16.495 |
3.751 |
T P |
| 2d61_A |
396 |
377 |
94 |
2.54 |
7.45 |
16.362 |
3.564 |
T P |
| 1aia_A |
396 |
377 |
97 |
2.86 |
8.25 |
16.061 |
3.273 |
T P |
| 1toi_A |
395 |
377 |
96 |
2.52 |
10.42 |
15.970 |
3.671 |
T P |
| 1bqa_A |
395 |
377 |
92 |
2.77 |
6.52 |
15.623 |
3.204 |
T P |
| 1aam_A |
396 |
377 |
94 |
2.58 |
13.83 |
15.578 |
3.502 |
T P |
| 1tok_A |
395 |
377 |
88 |
2.49 |
9.09 |
15.154 |
3.398 |
T P |
| 1qir_A |
396 |
377 |
92 |
2.75 |
7.61 |
15.144 |
3.226 |
T P |
| 1tog_A |
395 |
377 |
91 |
2.62 |
10.99 |
15.144 |
3.348 |
T P |
| 1arh_A |
396 |
377 |
90 |
2.81 |
7.78 |
15.104 |
3.098 |
T P |
| 4enl_A |
436 |
377 |
77 |
2.64 |
11.69 |
12.732 |
2.806 |
T P |
| 2al2_B |
430 |
377 |
71 |
2.69 |
8.45 |
12.402 |
2.543 |
T P |
| 1gkr_A |
450 |
377 |
75 |
2.76 |
9.33 |
12.125 |
2.627 |
T P |
| 2al1_A |
436 |
377 |
71 |
2.75 |
4.23 |
12.066 |
2.495 |
T P |
| 1l8p_A |
436 |
377 |
70 |
2.76 |
2.86 |
11.815 |
2.451 |
T P |
| 1p48_A |
436 |
377 |
68 |
2.67 |
4.41 |
11.712 |
2.453 |
T P |
| 1p43_A |
436 |
377 |
58 |
2.51 |
5.17 |
10.809 |
2.223 |
T P |
| 2al2_A |
436 |
377 |
62 |
2.93 |
9.68 |
10.630 |
2.043 |
T P |
| 1c5g_A |
379 |
377 |
59 |
2.53 |
5.08 |
10.412 |
2.244 |
T P |
| 5prn_A |
289 |
377 |
60 |
2.77 |
1.67 |
9.975 |
2.092 |
T P |
| 2iwk_A |
590 |
377 |
56 |
2.84 |
7.14 |
9.950 |
1.903 |
T P |
| 3prn_A |
289 |
377 |
59 |
2.76 |
8.47 |
9.903 |
2.062 |
T P |
| 3cvm_A |
377 |
377 |
51 |
2.60 |
3.92 |
9.686 |
1.891 |
T P |
| 1dvm_A |
375 |
377 |
53 |
2.68 |
11.32 |
9.338 |
1.906 |
T P |
| 3dc0_A |
422 |
377 |
51 |
2.62 |
3.92 |
9.306 |
1.874 |
T P |
| 2vzs_A |
857 |
377 |
52 |
2.79 |
7.69 |
9.212 |
1.801 |
T P |
| 1lj5_A |
379 |
377 |
49 |
2.62 |
6.12 |
8.847 |
1.803 |
T P |
| 1db2_A |
377 |
377 |
50 |
2.81 |
6.00 |
8.691 |
1.716 |
T P |
| 1bh3_A |
289 |
377 |
45 |
2.85 |
4.44 |
7.434 |
1.524 |
T P |
| 2prn_A |
289 |
377 |
41 |
2.84 |
4.88 |
7.126 |
1.393 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]