LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_323.5wLII_11276_25
Total number of 3D structures: 32
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
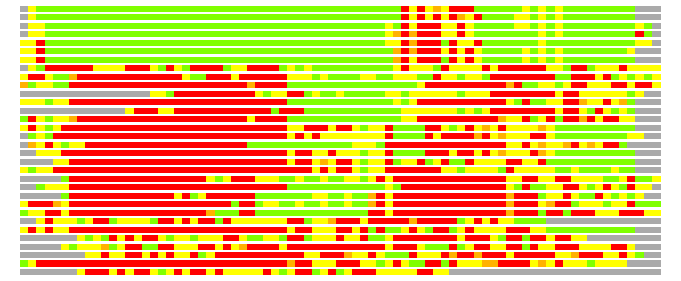
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1hr7_D |
441 |
79 |
70 |
1.35 |
14.29 |
85.954 |
4.844 |
T P |
| 1hr6_D |
443 |
79 |
70 |
1.39 |
14.29 |
85.693 |
4.692 |
T P |
| 1pp9_A |
442 |
79 |
72 |
1.50 |
23.61 |
85.399 |
4.495 |
T P |
| 1sqb_A |
446 |
79 |
71 |
1.53 |
23.94 |
85.043 |
4.367 |
T P |
| 1qcr_A |
446 |
79 |
71 |
1.54 |
23.94 |
84.749 |
4.331 |
T P |
| 1bcc_A |
442 |
79 |
69 |
1.47 |
21.74 |
83.866 |
4.390 |
T P |
| 3cwb_A |
443 |
79 |
69 |
1.49 |
21.74 |
83.139 |
4.331 |
T P |
| 1oyy_A |
512 |
79 |
48 |
2.51 |
6.25 |
40.930 |
1.841 |
T P |
| 1oyw_A |
516 |
79 |
42 |
2.30 |
9.52 |
38.392 |
1.747 |
T P |
| 1s2m_A |
377 |
79 |
36 |
2.35 |
8.33 |
35.387 |
1.472 |
T P |
| 2hyi_C |
392 |
79 |
39 |
2.47 |
7.69 |
34.437 |
1.520 |
T P |
| 2z0m_A |
331 |
79 |
34 |
2.21 |
8.82 |
33.909 |
1.472 |
T P |
| 2j0s_A |
391 |
79 |
34 |
2.19 |
0.00 |
33.753 |
1.487 |
T P |
| 2hxy_A |
376 |
79 |
33 |
2.29 |
3.03 |
32.808 |
1.382 |
T P |
| 1xti_A |
381 |
79 |
37 |
2.46 |
0.00 |
32.455 |
1.444 |
T P |
| 1t5i_A |
159 |
79 |
36 |
2.51 |
0.00 |
32.101 |
1.382 |
T P |
| 2zu6_C |
351 |
79 |
34 |
2.66 |
5.88 |
32.024 |
1.233 |
T P |
| 1xtk_A |
382 |
79 |
35 |
2.61 |
2.86 |
31.664 |
1.294 |
T P |
| 1fuk_A |
157 |
79 |
34 |
2.34 |
8.82 |
31.456 |
1.392 |
T P |
| 2rb4_A |
164 |
79 |
33 |
2.33 |
9.09 |
30.803 |
1.357 |
T P |
| 2vso_A |
366 |
79 |
34 |
2.49 |
5.88 |
30.620 |
1.312 |
T P |
| 2hjv_A |
158 |
79 |
31 |
2.03 |
9.68 |
30.584 |
1.454 |
T P |
| 3ews_A |
416 |
79 |
32 |
2.31 |
9.38 |
30.271 |
1.330 |
T P |
| 1fuu_B |
380 |
79 |
33 |
2.57 |
6.06 |
30.183 |
1.235 |
T P |
| 2i4i_A |
408 |
79 |
31 |
2.31 |
3.23 |
30.063 |
1.287 |
T P |
| 1hv8_A |
363 |
79 |
37 |
2.78 |
13.51 |
29.643 |
1.284 |
T P |
| 2g2j_A |
158 |
79 |
34 |
2.42 |
8.82 |
29.386 |
1.350 |
T P |
| 3fht_A |
392 |
79 |
36 |
2.68 |
8.33 |
28.685 |
1.295 |
T P |
| 2db3_A |
420 |
79 |
34 |
2.73 |
8.82 |
27.357 |
1.201 |
T P |
| 3g0h_A |
408 |
79 |
32 |
2.86 |
6.25 |
26.515 |
1.079 |
T P |
| 2p6n_A |
160 |
79 |
30 |
2.89 |
0.00 |
25.579 |
1.005 |
T P |
| 3eiq_D |
371 |
79 |
27 |
2.83 |
3.70 |
21.974 |
0.920 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]