LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_33.5wLII_10822_4
Total number of 3D structures: 74
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
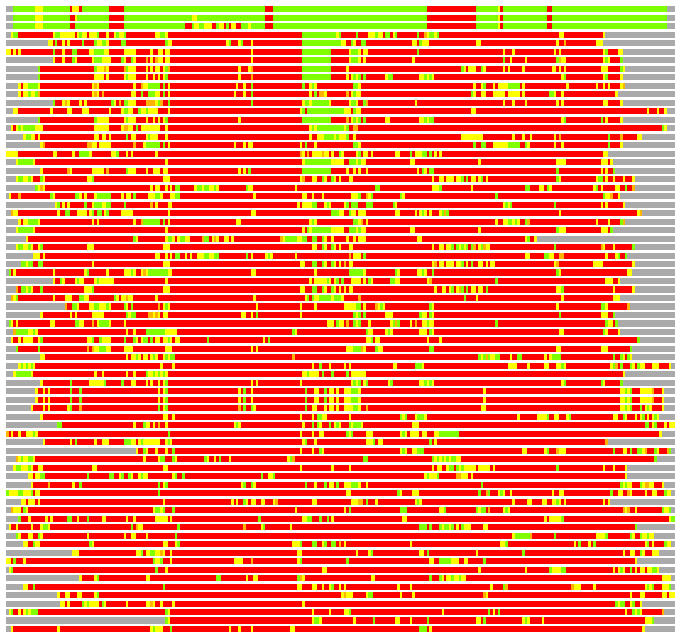
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2bh1_A |
238 |
273 |
233 |
0.83 |
18.88 |
84.018 |
24.974 |
T P |
| 1yf5_L |
238 |
273 |
233 |
1.04 |
18.45 |
82.471 |
20.364 |
T P |
| 1w97_L |
229 |
273 |
224 |
1.16 |
18.75 |
78.763 |
17.838 |
T P |
| 1dqw_A |
267 |
273 |
68 |
2.50 |
2.94 |
16.262 |
2.619 |
T P |
| 2f84_A |
323 |
273 |
57 |
2.36 |
12.28 |
15.257 |
2.313 |
T P |
| 1los_A |
216 |
273 |
55 |
2.54 |
0.00 |
13.955 |
2.082 |
T P |
| 1km4_A |
212 |
273 |
54 |
2.51 |
0.00 |
13.922 |
2.072 |
T P |
| 1km1_A |
212 |
273 |
54 |
2.66 |
3.70 |
13.917 |
1.958 |
T P |
| 1kly_A |
212 |
273 |
50 |
2.42 |
2.00 |
13.260 |
1.985 |
T P |
| 1lor_A |
209 |
273 |
52 |
2.56 |
5.77 |
13.217 |
1.954 |
T P |
| 1dv7_A |
212 |
273 |
55 |
2.83 |
5.45 |
13.208 |
1.875 |
T P |
| 1km5_A |
212 |
273 |
50 |
2.68 |
2.00 |
13.129 |
1.798 |
T P |
| 2yyu_B |
229 |
273 |
51 |
2.59 |
11.76 |
13.077 |
1.893 |
T P |
| 1km0_A |
237 |
273 |
49 |
2.37 |
2.04 |
12.990 |
1.984 |
T P |
| 1h1y_A |
219 |
273 |
53 |
2.53 |
7.55 |
12.980 |
2.012 |
T P |
| 2czd_B |
207 |
273 |
53 |
2.79 |
5.66 |
12.859 |
1.832 |
T P |
| 1km2_A |
211 |
273 |
51 |
2.58 |
5.88 |
12.843 |
1.900 |
T P |
| 2jgy_A |
249 |
273 |
50 |
2.86 |
4.00 |
12.667 |
1.689 |
T P |
| 1x1z_A |
215 |
273 |
44 |
2.45 |
4.55 |
12.137 |
1.726 |
T P |
| 1km3_A |
212 |
273 |
47 |
2.50 |
2.13 |
12.071 |
1.807 |
T P |
| 1ev1_1 |
281 |
273 |
50 |
2.80 |
18.00 |
11.841 |
1.727 |
T P |
| 3g3m_A |
257 |
273 |
45 |
2.60 |
4.44 |
11.616 |
1.667 |
T P |
| 2qce_A |
257 |
273 |
49 |
2.74 |
2.04 |
11.565 |
1.724 |
T P |
| 2p1f_A |
259 |
273 |
46 |
2.46 |
6.52 |
11.564 |
1.799 |
T P |
| 2guu_A |
317 |
273 |
49 |
2.78 |
6.12 |
11.542 |
1.702 |
T P |
| 1km6_A |
209 |
273 |
45 |
2.56 |
6.67 |
11.541 |
1.693 |
T P |
| 1loq_A |
209 |
273 |
43 |
2.37 |
6.98 |
11.486 |
1.744 |
T P |
| 1d4m_1 |
284 |
273 |
47 |
2.55 |
8.51 |
11.482 |
1.772 |
T P |
| 1rud_1 |
273 |
273 |
47 |
2.86 |
12.77 |
11.241 |
1.589 |
T P |
| 3g3d_B |
259 |
273 |
44 |
2.69 |
6.82 |
11.184 |
1.579 |
T P |
| 1rui_1 |
273 |
273 |
47 |
2.86 |
12.77 |
11.059 |
1.587 |
T P |
| 1eix_C |
232 |
273 |
45 |
2.54 |
2.22 |
10.949 |
1.703 |
T P |
| 3bk0_A |
257 |
273 |
45 |
2.75 |
2.22 |
10.890 |
1.579 |
T P |
| 1cov_1 |
269 |
273 |
45 |
2.74 |
24.44 |
10.847 |
1.585 |
T P |
| 2ffc_A |
318 |
273 |
42 |
2.59 |
2.38 |
10.762 |
1.561 |
T P |
| 1dbt_A |
237 |
273 |
42 |
2.70 |
7.14 |
10.731 |
1.499 |
T P |
| 1lol_B |
214 |
273 |
41 |
2.73 |
0.00 |
10.699 |
1.449 |
T P |
| 3bgg_A |
257 |
273 |
44 |
2.72 |
0.00 |
10.698 |
1.559 |
T P |
| 1klz_A |
209 |
273 |
43 |
2.48 |
0.00 |
10.691 |
1.664 |
T P |
| 1tqj_C |
218 |
273 |
42 |
2.67 |
2.38 |
10.585 |
1.517 |
T P |
| 1jjk_A |
231 |
273 |
39 |
2.73 |
5.13 |
10.499 |
1.378 |
T P |
| 3bpw_A |
330 |
273 |
41 |
2.53 |
4.88 |
10.299 |
1.558 |
T P |
| 1z7z_1 |
214 |
273 |
44 |
2.66 |
2.27 |
10.205 |
1.592 |
T P |
| 1tqx_A |
221 |
273 |
38 |
2.49 |
5.26 |
10.139 |
1.464 |
T P |
| 1dvj_A |
239 |
273 |
39 |
2.62 |
2.56 |
10.111 |
1.432 |
T P |
| 1pvc_1 |
279 |
273 |
38 |
2.64 |
2.63 |
9.923 |
1.386 |
T P |
| 3epd_1 |
279 |
273 |
38 |
2.64 |
2.63 |
9.923 |
1.386 |
T P |
| 1vbe_1 |
279 |
273 |
40 |
2.65 |
2.50 |
9.881 |
1.455 |
T P |
| 1hxs_1 |
288 |
273 |
40 |
2.66 |
5.00 |
9.842 |
1.449 |
T P |
| 1aym_1 |
285 |
273 |
36 |
2.65 |
5.56 |
9.779 |
1.310 |
T P |
| 1rue_1 |
273 |
273 |
39 |
2.64 |
2.56 |
9.707 |
1.424 |
T P |
| 1rpx_A |
230 |
273 |
36 |
2.75 |
2.78 |
9.705 |
1.262 |
T P |
| 1oop_A |
271 |
273 |
38 |
2.59 |
2.63 |
9.695 |
1.411 |
T P |
| 1mqt_A |
271 |
273 |
37 |
2.51 |
5.41 |
9.654 |
1.420 |
T P |
| 1ncq_A |
273 |
273 |
41 |
2.91 |
7.32 |
9.451 |
1.362 |
T P |
| 1al2_1 |
283 |
273 |
37 |
2.53 |
0.00 |
9.387 |
1.406 |
T P |
| 1vbb_1 |
279 |
273 |
38 |
2.64 |
2.63 |
9.385 |
1.387 |
T P |
| 1bev_1 |
268 |
273 |
42 |
2.79 |
4.76 |
9.377 |
1.453 |
T P |
| 2qcf_A |
257 |
273 |
41 |
2.85 |
12.20 |
9.294 |
1.389 |
T P |
| 1pov_1 |
235 |
273 |
37 |
2.61 |
2.70 |
9.269 |
1.364 |
T P |
| 1z7s_1 |
283 |
273 |
37 |
2.59 |
8.11 |
9.241 |
1.374 |
T P |
| 1rug_1 |
273 |
273 |
34 |
2.62 |
8.82 |
8.832 |
1.249 |
T P |
| 1fpn_1 |
269 |
273 |
34 |
2.57 |
11.76 |
8.663 |
1.275 |
T P |
| 1ar7_1 |
283 |
273 |
37 |
2.72 |
10.81 |
8.562 |
1.312 |
T P |
| 1r1a_1 |
283 |
273 |
33 |
2.81 |
15.15 |
8.337 |
1.133 |
T P |
| 2rmu_1 |
273 |
273 |
31 |
2.68 |
12.90 |
7.991 |
1.114 |
T P |
| 1h8t_A |
289 |
273 |
28 |
2.76 |
3.57 |
7.578 |
0.980 |
T P |
| 3epc_1 |
283 |
273 |
30 |
2.84 |
6.67 |
7.542 |
1.022 |
T P |
| 1ar6_1 |
283 |
273 |
31 |
2.68 |
16.13 |
7.445 |
1.115 |
T P |
| 1eah_1 |
272 |
273 |
26 |
3.03 |
3.85 |
7.107 |
0.831 |
T P |
| 3epf_1 |
272 |
273 |
27 |
3.04 |
7.41 |
6.655 |
0.860 |
T P |
| 1rhi_1 |
273 |
273 |
26 |
2.52 |
3.85 |
6.619 |
0.992 |
T P |
| 1z8r_A |
150 |
273 |
23 |
2.76 |
8.70 |
5.863 |
0.805 |
T P |
| 1rmu_1 |
273 |
273 |
18 |
2.53 |
5.56 |
4.821 |
0.686 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]