LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_331.5wLII_11276_77
Total number of 3D structures: 88
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
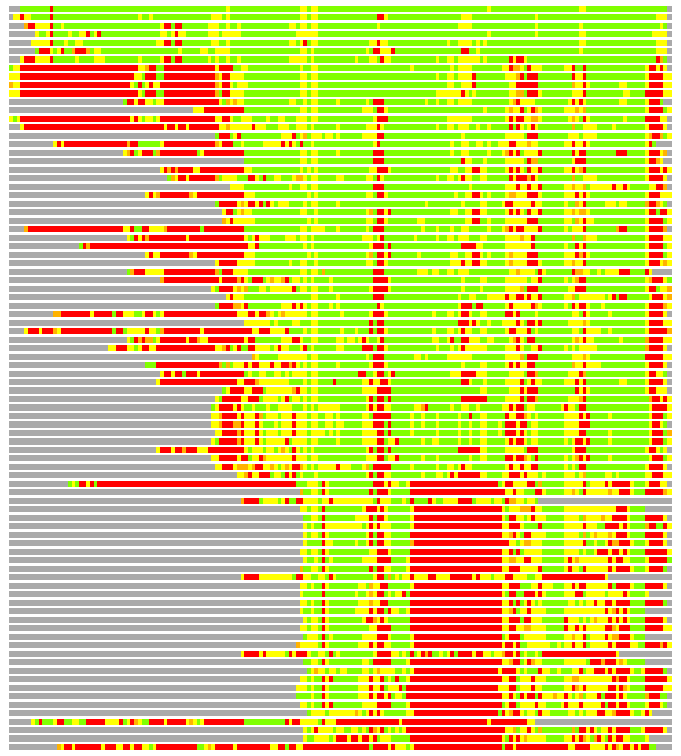
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2yxe_A |
214 |
180 |
175 |
0.73 |
17.71 |
96.294 |
20.979 |
T P |
| 1jg1_A |
215 |
180 |
174 |
1.28 |
13.22 |
91.910 |
12.591 |
T P |
| 1i1n_A |
224 |
180 |
168 |
1.49 |
11.31 |
86.252 |
10.536 |
T P |
| 1dl5_A |
317 |
180 |
168 |
1.58 |
14.88 |
85.931 |
9.995 |
T P |
| 1r18_A |
223 |
180 |
166 |
1.58 |
12.65 |
85.022 |
9.872 |
T P |
| 1vbf_B |
226 |
180 |
160 |
1.47 |
12.50 |
83.438 |
10.199 |
T P |
| 2pbf_A |
218 |
180 |
163 |
1.70 |
11.66 |
82.417 |
9.060 |
T P |
| 1o54_A |
265 |
180 |
120 |
1.79 |
7.50 |
60.425 |
6.336 |
T P |
| 1i9g_A |
264 |
180 |
119 |
1.78 |
14.29 |
60.028 |
6.317 |
T P |
| 2b25_A |
254 |
180 |
119 |
1.86 |
10.08 |
59.631 |
6.079 |
T P |
| 2pwy_A |
251 |
180 |
119 |
1.86 |
12.61 |
58.963 |
6.058 |
T P |
| 1l3i_A |
186 |
180 |
119 |
1.94 |
9.24 |
58.661 |
5.822 |
T P |
| 3bus_A |
251 |
180 |
111 |
1.84 |
13.51 |
56.405 |
5.728 |
T P |
| 2yvl_A |
247 |
180 |
120 |
2.04 |
5.83 |
56.234 |
5.614 |
T P |
| 1t43_A |
274 |
180 |
117 |
2.10 |
10.26 |
54.833 |
5.322 |
T P |
| 3e7p_A |
253 |
180 |
112 |
1.94 |
8.93 |
54.654 |
5.504 |
T P |
| 3duw_A |
220 |
180 |
118 |
1.96 |
12.71 |
54.272 |
5.735 |
T P |
| 2bh2_A |
418 |
180 |
108 |
1.79 |
12.04 |
54.159 |
5.718 |
T P |
| 1xxl_A |
234 |
180 |
104 |
1.72 |
8.65 |
54.154 |
5.701 |
T P |
| 1kph_B |
285 |
180 |
106 |
1.80 |
11.32 |
54.008 |
5.591 |
T P |
| 2b3t_A |
276 |
180 |
113 |
2.03 |
8.85 |
53.992 |
5.306 |
T P |
| 2h1r_B |
275 |
180 |
109 |
1.92 |
11.01 |
53.776 |
5.390 |
T P |
| 1tpy_A |
285 |
180 |
109 |
1.96 |
12.84 |
53.453 |
5.298 |
T P |
| 3dh0_B |
190 |
180 |
108 |
1.98 |
10.19 |
53.248 |
5.185 |
T P |
| 1kpg_A |
285 |
180 |
107 |
1.86 |
12.15 |
53.092 |
5.465 |
T P |
| 1kpi_A |
291 |
180 |
109 |
1.92 |
11.01 |
52.907 |
5.408 |
T P |
| 1uwv_A |
417 |
180 |
112 |
2.04 |
13.39 |
52.609 |
5.227 |
T P |
| 1sqg_A |
424 |
180 |
114 |
2.12 |
9.65 |
52.484 |
5.131 |
T P |
| 3ccf_B |
242 |
180 |
101 |
1.66 |
6.93 |
52.417 |
5.728 |
T P |
| 1l1e_A |
272 |
180 |
107 |
1.96 |
12.15 |
52.352 |
5.201 |
T P |
| 2fk8_A |
281 |
180 |
107 |
1.95 |
12.15 |
51.987 |
5.223 |
T P |
| 2yxd_A |
180 |
180 |
114 |
2.16 |
3.51 |
51.711 |
5.046 |
T P |
| 3d2l_C |
242 |
180 |
105 |
1.98 |
11.43 |
51.570 |
5.056 |
T P |
| 1ve3_B |
226 |
180 |
108 |
1.98 |
9.26 |
51.535 |
5.193 |
T P |
| 1zq9_A |
278 |
180 |
106 |
1.91 |
16.04 |
51.029 |
5.271 |
T P |
| 1p91_A |
268 |
180 |
102 |
1.86 |
11.76 |
50.988 |
5.203 |
T P |
| 3cjt_A |
254 |
180 |
113 |
2.09 |
11.50 |
50.910 |
5.161 |
T P |
| 3bkw_A |
219 |
180 |
101 |
1.87 |
7.92 |
50.614 |
5.132 |
T P |
| 2nxc_A |
249 |
180 |
113 |
2.15 |
12.39 |
50.419 |
5.022 |
T P |
| 3dtn_A |
220 |
180 |
110 |
2.17 |
8.18 |
50.041 |
4.851 |
T P |
| 1dus_A |
194 |
180 |
113 |
2.12 |
10.62 |
49.816 |
5.091 |
T P |
| 1vl5_A |
231 |
180 |
103 |
1.96 |
6.80 |
49.383 |
4.996 |
T P |
| 3dmg_A |
371 |
180 |
106 |
2.01 |
16.04 |
48.955 |
5.017 |
T P |
| 2gs9_A |
211 |
180 |
105 |
2.03 |
11.43 |
48.478 |
4.927 |
T P |
| 2pxx_A |
213 |
180 |
101 |
2.06 |
13.86 |
48.151 |
4.673 |
T P |
| 1wzn_A |
244 |
180 |
101 |
1.95 |
6.93 |
48.072 |
4.923 |
T P |
| 3e23_A |
198 |
180 |
97 |
1.88 |
7.22 |
47.142 |
4.896 |
T P |
| 1nkv_C |
249 |
180 |
106 |
2.04 |
9.43 |
44.271 |
4.945 |
T P |
| 1r74_B |
279 |
180 |
104 |
2.13 |
7.69 |
43.893 |
4.659 |
T P |
| 1d2g_A |
292 |
180 |
104 |
2.15 |
8.65 |
43.838 |
4.617 |
T P |
| 1r8x_B |
284 |
180 |
105 |
2.15 |
8.57 |
43.713 |
4.665 |
T P |
| 1xva_A |
292 |
180 |
105 |
2.11 |
5.71 |
43.499 |
4.748 |
T P |
| 2avn_A |
247 |
180 |
102 |
2.00 |
16.67 |
43.278 |
4.867 |
T P |
| 2azt_B |
277 |
180 |
103 |
2.13 |
5.83 |
43.001 |
4.621 |
T P |
| 2yqz_A |
261 |
180 |
99 |
2.31 |
11.11 |
42.728 |
4.101 |
T P |
| 3cc8_A |
211 |
180 |
95 |
2.13 |
9.47 |
39.410 |
4.267 |
T P |
| 1w25_A |
454 |
180 |
63 |
2.29 |
11.11 |
26.196 |
2.636 |
T P |
| 1xhf_B |
122 |
180 |
58 |
2.34 |
5.17 |
24.830 |
2.375 |
T P |
| 2ftk_E |
119 |
180 |
65 |
2.57 |
10.77 |
24.786 |
2.439 |
T P |
| 1kgs_A |
219 |
180 |
61 |
2.50 |
4.92 |
23.150 |
2.345 |
T P |
| 2jba_A |
125 |
180 |
59 |
2.41 |
10.17 |
23.058 |
2.348 |
T P |
| 2oqr_A |
226 |
180 |
60 |
2.36 |
10.00 |
23.001 |
2.437 |
T P |
| 1zh2_A |
120 |
180 |
59 |
2.39 |
16.95 |
22.985 |
2.370 |
T P |
| 1f51_E |
119 |
180 |
60 |
2.37 |
5.00 |
22.893 |
2.431 |
T P |
| 1zes_A |
121 |
180 |
58 |
2.34 |
8.62 |
22.748 |
2.374 |
T P |
| 1nat_A |
119 |
180 |
59 |
2.41 |
6.78 |
22.705 |
2.351 |
T P |
| 1xhe_B |
122 |
180 |
56 |
2.20 |
7.14 |
22.647 |
2.434 |
T P |
| 2jvi_A |
132 |
180 |
62 |
2.60 |
8.06 |
22.524 |
2.294 |
T P |
| 2j8e_A |
128 |
180 |
56 |
2.29 |
7.14 |
22.480 |
2.341 |
T P |
| 1srr_C |
121 |
180 |
57 |
2.19 |
3.51 |
22.457 |
2.490 |
T P |
| 2fka_A |
128 |
180 |
57 |
2.28 |
8.77 |
22.420 |
2.395 |
T P |
| 3c3m_A |
123 |
180 |
59 |
2.39 |
10.17 |
22.161 |
2.370 |
T P |
| 2a9r_A |
117 |
180 |
56 |
2.41 |
8.93 |
22.030 |
2.232 |
T P |
| 2a9o_A |
117 |
180 |
56 |
2.33 |
8.93 |
22.002 |
2.308 |
T P |
| 2jba_B |
121 |
180 |
57 |
2.37 |
8.77 |
21.876 |
2.311 |
T P |
| 2chy_A |
128 |
180 |
59 |
2.50 |
10.17 |
21.765 |
2.269 |
T P |
| 2jvk_A |
132 |
180 |
56 |
2.44 |
8.93 |
21.750 |
2.208 |
T P |
| 1b00_A |
122 |
180 |
53 |
2.34 |
9.43 |
21.617 |
2.170 |
T P |
| 1zgz_A |
121 |
180 |
56 |
2.26 |
12.50 |
21.537 |
2.377 |
T P |
| 1ab5_A |
125 |
180 |
59 |
2.57 |
6.78 |
21.467 |
2.210 |
T P |
| 2che_A |
128 |
180 |
60 |
2.58 |
6.67 |
21.290 |
2.243 |
T P |
| 2iyn_B |
124 |
180 |
56 |
2.42 |
7.14 |
21.209 |
2.219 |
T P |
| 1mvo_A |
121 |
180 |
56 |
2.37 |
8.93 |
20.975 |
2.263 |
T P |
| 2jb9_B |
122 |
180 |
58 |
2.53 |
10.34 |
20.849 |
2.208 |
T P |
| 2gwr_A |
225 |
180 |
53 |
2.49 |
9.43 |
20.697 |
2.045 |
T P |
| 1ab6_A |
125 |
180 |
55 |
2.56 |
3.64 |
20.159 |
2.071 |
T P |
| 2jvj_A |
132 |
180 |
53 |
2.51 |
7.55 |
19.266 |
2.030 |
T P |
| 1ys7_B |
226 |
180 |
44 |
2.97 |
11.36 |
15.837 |
1.431 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]