LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_333.5wLII_11276_87
Total number of 3D structures: 59
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
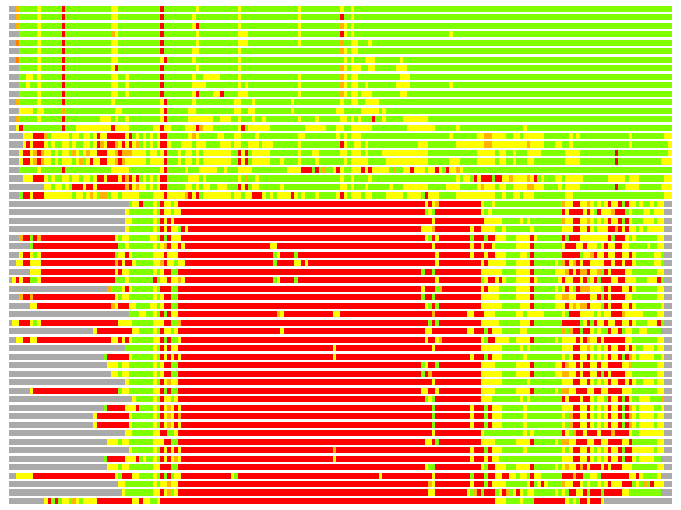
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3bjf_A |
518 |
188 |
184 |
1.05 |
17.93 |
95.705 |
16.046 |
T P |
| 1t5a_A |
519 |
188 |
183 |
1.02 |
18.03 |
95.416 |
16.402 |
T P |
| 1pkm_A |
519 |
188 |
183 |
1.17 |
18.03 |
94.448 |
14.393 |
T P |
| 3g2g_C |
510 |
188 |
182 |
1.11 |
18.13 |
94.424 |
15.034 |
T P |
| 1f3w_A |
519 |
188 |
184 |
1.25 |
17.93 |
94.060 |
13.637 |
T P |
| 3bjt_A |
512 |
188 |
183 |
1.17 |
18.03 |
93.988 |
14.355 |
T P |
| 1lix_A |
517 |
188 |
184 |
1.27 |
15.22 |
93.752 |
13.456 |
T P |
| 1liy_C |
514 |
188 |
182 |
1.23 |
15.38 |
93.162 |
13.652 |
T P |
| 1f3x_A |
519 |
188 |
183 |
1.28 |
18.03 |
93.076 |
13.249 |
T P |
| 2g50_D |
521 |
188 |
183 |
1.33 |
18.03 |
92.374 |
12.837 |
T P |
| 1pkn_A |
514 |
188 |
181 |
1.28 |
17.13 |
91.919 |
13.119 |
T P |
| 1liu_A |
517 |
188 |
184 |
1.46 |
15.22 |
91.758 |
11.824 |
T P |
| 1liw_A |
517 |
188 |
184 |
1.51 |
15.22 |
90.936 |
11.431 |
T P |
| 1a3w_A |
492 |
188 |
183 |
1.60 |
15.30 |
90.073 |
10.746 |
T P |
| 1a49_A |
519 |
188 |
180 |
1.88 |
17.78 |
82.168 |
9.081 |
T P |
| 1zjh_A |
507 |
188 |
172 |
2.07 |
15.70 |
75.959 |
7.927 |
T P |
| 1pkl_G |
498 |
188 |
172 |
2.18 |
12.21 |
74.138 |
7.531 |
T P |
| 1pky_A |
464 |
188 |
174 |
2.31 |
13.22 |
73.988 |
7.232 |
T P |
| 1e0u_A |
461 |
188 |
174 |
2.30 |
13.79 |
73.325 |
7.248 |
T P |
| 3e0w_A |
485 |
188 |
173 |
1.88 |
12.72 |
72.180 |
8.724 |
T P |
| 2e28_A |
587 |
188 |
164 |
2.03 |
14.02 |
68.973 |
7.694 |
T P |
| 1e0t_A |
446 |
188 |
159 |
2.20 |
11.95 |
64.880 |
6.909 |
T P |
| 3eoe_B |
483 |
188 |
171 |
2.32 |
14.04 |
64.755 |
7.073 |
T P |
| 2g4r_A |
153 |
188 |
62 |
2.31 |
11.29 |
24.426 |
2.569 |
T P |
| 2g2c_A |
152 |
188 |
60 |
2.18 |
6.67 |
24.283 |
2.634 |
T P |
| 1di6_A |
183 |
188 |
59 |
2.27 |
10.17 |
23.671 |
2.489 |
T P |
| 2pjk_A |
166 |
188 |
65 |
2.47 |
9.23 |
23.527 |
2.531 |
T P |
| 1t3e_A |
412 |
188 |
64 |
2.39 |
9.38 |
23.467 |
2.573 |
T P |
| 1uz5_A |
391 |
188 |
64 |
2.41 |
6.25 |
23.360 |
2.548 |
T P |
| 1fc5_A |
404 |
188 |
63 |
2.39 |
4.76 |
23.244 |
2.530 |
T P |
| 2nqv_A |
405 |
188 |
66 |
2.66 |
4.55 |
23.208 |
2.395 |
T P |
| 1g8l_A |
403 |
188 |
63 |
2.49 |
3.17 |
23.190 |
2.436 |
T P |
| 2fts_A |
419 |
188 |
64 |
2.34 |
7.81 |
23.121 |
2.627 |
T P |
| 2nqq_A |
403 |
188 |
62 |
2.47 |
6.45 |
23.091 |
2.416 |
T P |
| 2nqk_A |
403 |
188 |
61 |
2.37 |
4.92 |
23.064 |
2.470 |
T P |
| 2nqm_B |
403 |
188 |
63 |
2.46 |
6.35 |
23.027 |
2.463 |
T P |
| 2is8_B |
161 |
188 |
64 |
2.58 |
9.38 |
22.952 |
2.385 |
T P |
| 2nrp_B |
405 |
188 |
62 |
2.26 |
4.84 |
22.947 |
2.625 |
T P |
| 2f7w_A |
173 |
188 |
61 |
2.28 |
8.20 |
22.930 |
2.568 |
T P |
| 2nqu_B |
404 |
188 |
63 |
2.48 |
4.76 |
22.880 |
2.441 |
T P |
| 1jlj_A |
169 |
188 |
61 |
2.33 |
6.56 |
22.839 |
2.509 |
T P |
| 1eav_A |
160 |
188 |
59 |
2.28 |
8.47 |
22.787 |
2.476 |
T P |
| 2nqs_A |
403 |
188 |
63 |
2.37 |
4.76 |
22.720 |
2.555 |
T P |
| 2nro_A |
403 |
188 |
61 |
2.31 |
6.56 |
22.694 |
2.534 |
T P |
| 1ihc_A |
169 |
188 |
60 |
2.31 |
6.67 |
22.670 |
2.489 |
T P |
| 2nqn_A |
405 |
188 |
61 |
2.35 |
3.28 |
22.614 |
2.493 |
T P |
| 2pbq_A |
174 |
188 |
58 |
2.14 |
6.90 |
22.584 |
2.584 |
T P |
| 1o8n_A |
162 |
188 |
60 |
2.37 |
8.33 |
22.465 |
2.429 |
T P |
| 1uuy_A |
161 |
188 |
60 |
2.35 |
6.67 |
22.457 |
2.454 |
T P |
| 1uux_A |
161 |
188 |
60 |
2.34 |
6.67 |
22.457 |
2.457 |
T P |
| 1mkz_B |
171 |
188 |
53 |
2.13 |
3.77 |
22.413 |
2.374 |
T P |
| 2nqr_A |
404 |
188 |
62 |
2.33 |
4.84 |
22.326 |
2.546 |
T P |
| 1o8q_A |
159 |
188 |
57 |
2.27 |
7.02 |
22.303 |
2.403 |
T P |
| 1o8o_B |
163 |
188 |
58 |
2.35 |
8.62 |
22.273 |
2.370 |
T P |
| 2nrs_A |
403 |
188 |
60 |
2.35 |
5.00 |
21.836 |
2.453 |
T P |
| 1wu2_A |
383 |
188 |
55 |
2.37 |
5.45 |
21.350 |
2.228 |
T P |
| 1y5e_C |
161 |
188 |
61 |
2.53 |
6.56 |
20.957 |
2.318 |
T P |
| 1r2k_B |
168 |
188 |
55 |
2.29 |
3.64 |
20.504 |
2.301 |
T P |
| 3bfj_A |
382 |
188 |
37 |
2.36 |
2.70 |
14.605 |
1.507 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]