LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_336.5wLII_11276_103
Total number of 3D structures: 59
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
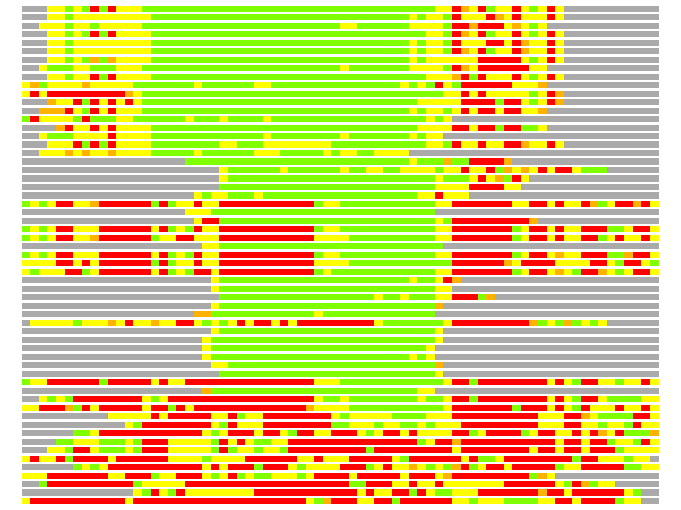
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1a1i_A |
85 |
74 |
54 |
1.95 |
11.11 |
66.476 |
2.634 |
T P |
| 1aay_A |
85 |
74 |
56 |
2.14 |
14.29 |
64.884 |
2.496 |
T P |
| 1g2f_C |
89 |
74 |
54 |
2.06 |
12.96 |
64.862 |
2.505 |
T P |
| 1a1g_A |
84 |
74 |
54 |
1.97 |
11.11 |
64.681 |
2.613 |
T P |
| 1zaa_C |
85 |
74 |
55 |
2.08 |
14.55 |
64.423 |
2.523 |
T P |
| 1a1h_A |
85 |
74 |
56 |
2.15 |
12.50 |
64.245 |
2.489 |
T P |
| 1jk2_A |
85 |
74 |
54 |
2.16 |
11.11 |
63.937 |
2.384 |
T P |
| 1g2d_C |
89 |
74 |
52 |
2.00 |
13.46 |
63.905 |
2.482 |
T P |
| 1p47_A |
87 |
74 |
54 |
2.00 |
11.11 |
63.878 |
2.574 |
T P |
| 2gli_A |
155 |
74 |
54 |
2.16 |
16.67 |
63.210 |
2.385 |
T P |
| 2i13_A |
151 |
74 |
50 |
1.77 |
14.00 |
63.155 |
2.679 |
T P |
| 2prt_A |
115 |
74 |
49 |
1.90 |
12.24 |
63.047 |
2.451 |
T P |
| 1mey_F |
84 |
74 |
51 |
2.12 |
9.80 |
62.207 |
2.298 |
T P |
| 1tf3_A |
92 |
74 |
48 |
1.78 |
8.33 |
60.260 |
2.553 |
T P |
| 1ubd_C |
114 |
74 |
48 |
1.84 |
6.25 |
59.134 |
2.468 |
T P |
| 1llm_C |
87 |
74 |
46 |
2.03 |
15.22 |
56.312 |
2.162 |
T P |
| 1tf6_D |
182 |
74 |
52 |
2.29 |
13.46 |
55.659 |
2.178 |
T P |
| 1f2i_G |
66 |
74 |
43 |
2.41 |
9.30 |
50.396 |
1.711 |
T P |
| 1x6e_A |
72 |
74 |
34 |
1.71 |
17.65 |
44.470 |
1.877 |
T P |
| 2dlq_A |
124 |
74 |
40 |
2.26 |
7.50 |
44.002 |
1.695 |
T P |
| 2eoe_A |
46 |
74 |
34 |
1.77 |
17.65 |
43.123 |
1.819 |
T P |
| 2ee8_A |
106 |
74 |
31 |
1.74 |
12.90 |
39.581 |
1.683 |
T P |
| 2ytb_A |
42 |
74 |
31 |
1.80 |
9.68 |
39.385 |
1.628 |
T P |
| 1m2j_A |
249 |
74 |
39 |
2.40 |
5.13 |
39.347 |
1.561 |
T P |
| 2eok_A |
42 |
74 |
29 |
1.20 |
20.69 |
38.946 |
2.237 |
T P |
| 2ytr_A |
46 |
74 |
29 |
1.55 |
17.24 |
38.573 |
1.763 |
T P |
| 1m2g_A |
249 |
74 |
38 |
2.30 |
5.26 |
38.146 |
1.583 |
T P |
| 1m2h_A |
249 |
74 |
39 |
2.43 |
2.56 |
37.951 |
1.542 |
T P |
| 2yu8_A |
46 |
74 |
28 |
1.03 |
14.29 |
37.885 |
2.478 |
T P |
| 1m2k_A |
249 |
74 |
39 |
2.45 |
5.13 |
37.811 |
1.529 |
T P |
| 1ici_A |
256 |
74 |
38 |
2.55 |
7.89 |
37.633 |
1.434 |
T P |
| 1m2n_A |
249 |
74 |
39 |
2.38 |
7.69 |
37.558 |
1.570 |
T P |
| 2yt9_A |
95 |
74 |
28 |
1.41 |
17.86 |
37.364 |
1.860 |
T P |
| 2yts_A |
46 |
74 |
28 |
1.20 |
14.29 |
37.192 |
2.153 |
T P |
| 2eq0_A |
46 |
74 |
29 |
1.62 |
17.24 |
36.788 |
1.688 |
T P |
| 2em2_A |
46 |
74 |
27 |
1.07 |
14.81 |
36.718 |
2.302 |
T P |
| 2ep1_A |
46 |
74 |
28 |
1.70 |
17.86 |
36.617 |
1.555 |
T P |
| 2be5_D |
1392 |
74 |
42 |
2.70 |
7.14 |
36.506 |
1.501 |
T P |
| 2ytk_A |
46 |
74 |
27 |
1.09 |
11.11 |
36.409 |
2.264 |
T P |
| 2emw_A |
44 |
74 |
28 |
1.29 |
17.86 |
36.326 |
2.009 |
T P |
| 2emv_A |
44 |
74 |
27 |
1.09 |
18.52 |
36.279 |
2.276 |
T P |
| 2adr_A |
60 |
74 |
27 |
1.17 |
14.81 |
36.190 |
2.131 |
T P |
| 2enf_A |
46 |
74 |
27 |
1.52 |
14.81 |
35.123 |
1.662 |
T P |
| 2ene_A |
46 |
74 |
26 |
1.04 |
19.23 |
35.028 |
2.290 |
T P |
| 2h59_A |
246 |
74 |
33 |
2.12 |
9.09 |
34.626 |
1.486 |
T P |
| 2cot_A |
77 |
74 |
27 |
1.53 |
11.11 |
34.382 |
1.658 |
T P |
| 1yc5_A |
234 |
74 |
34 |
2.18 |
5.88 |
34.306 |
1.490 |
T P |
| 2h4h_A |
237 |
74 |
35 |
2.48 |
8.57 |
33.086 |
1.355 |
T P |
| 2ot4_A |
520 |
74 |
35 |
2.69 |
0.00 |
32.241 |
1.255 |
T P |
| 3f29_B |
520 |
74 |
33 |
2.47 |
3.03 |
32.173 |
1.285 |
T P |
| 1h3n_A |
814 |
74 |
32 |
2.66 |
12.50 |
30.118 |
1.160 |
T P |
| 1dgt_B |
660 |
74 |
32 |
2.55 |
6.25 |
29.922 |
1.208 |
T P |
| 1v9p_A |
584 |
74 |
32 |
2.77 |
3.12 |
29.559 |
1.114 |
T P |
| 3f2b_A |
994 |
74 |
32 |
2.66 |
9.38 |
28.841 |
1.161 |
T P |
| 2v0c_A |
810 |
74 |
33 |
2.90 |
12.12 |
28.443 |
1.101 |
T P |
| 1xx6_A |
178 |
74 |
27 |
2.68 |
18.52 |
25.450 |
0.971 |
T P |
| 2csh_A |
110 |
74 |
26 |
2.76 |
19.23 |
24.501 |
0.909 |
T P |
| 1dfx_A |
125 |
74 |
26 |
2.64 |
0.00 |
23.996 |
0.947 |
T P |
| 2ba1_A |
179 |
74 |
24 |
2.61 |
4.17 |
23.976 |
0.887 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]