LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_35.5wLII_10822_10
Total number of 3D structures: 63
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
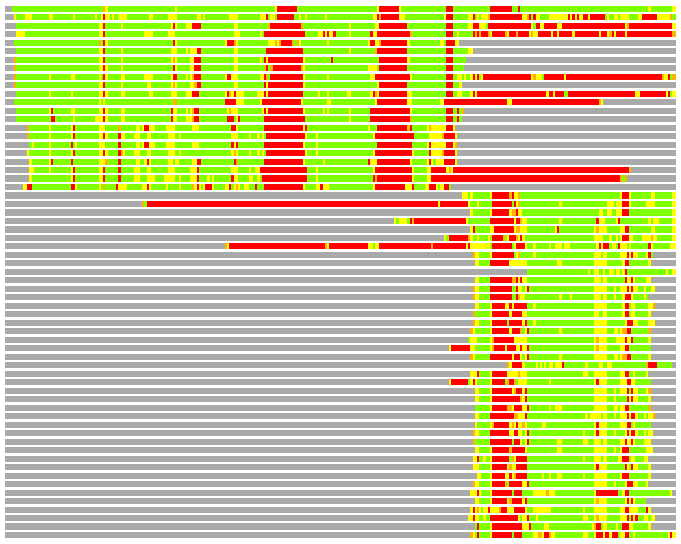
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2z9i_A |
284 |
308 |
273 |
0.67 |
16.48 |
87.832 |
35.386 |
T P |
| 3cs0_A |
392 |
308 |
245 |
2.05 |
10.61 |
68.821 |
11.399 |
T P |
| 1soz_A |
281 |
308 |
187 |
1.47 |
10.16 |
58.656 |
11.920 |
T P |
| 1ky9_B |
396 |
308 |
188 |
1.87 |
10.64 |
55.687 |
9.522 |
T P |
| 1l1j_A |
226 |
308 |
177 |
1.58 |
9.60 |
54.745 |
10.532 |
T P |
| 2qf3_A |
200 |
308 |
173 |
1.21 |
9.83 |
54.093 |
13.159 |
T P |
| 2rce_C |
213 |
308 |
170 |
1.20 |
10.00 |
54.004 |
13.070 |
T P |
| 2qf0_B |
211 |
308 |
172 |
1.39 |
9.88 |
53.937 |
11.570 |
T P |
| 1sot_B |
294 |
308 |
183 |
1.82 |
8.74 |
53.520 |
9.550 |
T P |
| 2r3u_A |
202 |
308 |
170 |
1.33 |
10.00 |
52.781 |
11.924 |
T P |
| 1te0_A |
318 |
308 |
180 |
1.77 |
9.44 |
52.040 |
9.606 |
T P |
| 1lcy_A |
296 |
308 |
165 |
1.48 |
10.91 |
50.834 |
10.444 |
T P |
| 3b8j_A |
184 |
308 |
162 |
1.54 |
11.11 |
49.509 |
9.904 |
T P |
| 2qgr_A |
169 |
308 |
154 |
1.36 |
11.04 |
47.558 |
10.546 |
T P |
| 2o8l_A |
216 |
308 |
155 |
1.73 |
7.10 |
46.567 |
8.454 |
T P |
| 1agj_A |
242 |
308 |
154 |
1.71 |
8.44 |
46.239 |
8.494 |
T P |
| 1wcz_A |
215 |
308 |
153 |
1.70 |
8.50 |
46.236 |
8.497 |
T P |
| 1due_A |
242 |
308 |
154 |
1.73 |
8.44 |
46.128 |
8.397 |
T P |
| 2as9_A |
207 |
308 |
150 |
1.69 |
9.33 |
45.550 |
8.377 |
T P |
| 1qtf_A |
246 |
308 |
146 |
1.78 |
5.48 |
43.586 |
7.762 |
T P |
| 1dt2_A |
245 |
308 |
145 |
1.79 |
6.21 |
43.262 |
7.676 |
T P |
| 2w5e_A |
162 |
308 |
146 |
1.86 |
6.16 |
42.723 |
7.450 |
T P |
| 2p3w_A |
110 |
308 |
86 |
1.56 |
18.60 |
26.202 |
5.186 |
T P |
| 2ytw_A |
118 |
308 |
85 |
1.73 |
16.47 |
25.504 |
4.652 |
T P |
| 2pzd_A |
106 |
308 |
82 |
1.60 |
23.17 |
24.973 |
4.827 |
T P |
| 1fc9_A |
386 |
308 |
89 |
1.95 |
19.10 |
24.157 |
4.340 |
T P |
| 2qbw_A |
189 |
308 |
81 |
1.98 |
22.22 |
23.532 |
3.892 |
T P |
| 2joa_A |
105 |
308 |
85 |
2.09 |
17.65 |
23.292 |
3.874 |
T P |
| 1fc6_A |
386 |
308 |
86 |
2.27 |
19.77 |
21.355 |
3.623 |
T P |
| 2d90_A |
102 |
308 |
70 |
1.82 |
20.00 |
20.568 |
3.638 |
T P |
| 1ihj_B |
95 |
308 |
70 |
1.82 |
18.57 |
20.476 |
3.648 |
T P |
| 2zpm_A |
86 |
308 |
67 |
1.48 |
22.39 |
20.400 |
4.238 |
T P |
| 1g9o_A |
91 |
308 |
67 |
1.72 |
20.90 |
20.371 |
3.676 |
T P |
| 1gq4_A |
90 |
308 |
70 |
1.89 |
18.57 |
20.258 |
3.526 |
T P |
| 1x5n_A |
114 |
308 |
67 |
1.70 |
26.87 |
20.202 |
3.732 |
T P |
| 1gq5_A |
91 |
308 |
68 |
1.85 |
19.12 |
20.100 |
3.494 |
T P |
| 2f5y_A |
82 |
308 |
67 |
1.64 |
13.43 |
19.926 |
3.842 |
T P |
| 1i92_A |
91 |
308 |
67 |
1.79 |
19.40 |
19.855 |
3.545 |
T P |
| 1be9_A |
115 |
308 |
68 |
2.05 |
22.06 |
19.775 |
3.167 |
T P |
| 3dj3_B |
101 |
308 |
73 |
1.94 |
20.55 |
19.767 |
3.580 |
T P |
| 2vz5_A |
111 |
308 |
71 |
1.82 |
19.72 |
19.714 |
3.703 |
T P |
| 1tp5_A |
115 |
308 |
67 |
2.00 |
22.39 |
19.704 |
3.186 |
T P |
| 2hga_A |
103 |
308 |
66 |
1.83 |
31.82 |
19.656 |
3.426 |
T P |
| 1x5q_A |
110 |
308 |
72 |
2.02 |
18.06 |
19.550 |
3.395 |
T P |
| 3dj1_A |
115 |
308 |
73 |
2.01 |
20.55 |
19.452 |
3.460 |
T P |
| 2v90_A |
95 |
308 |
66 |
1.84 |
19.70 |
19.435 |
3.405 |
T P |
| 2ocs_A |
85 |
308 |
65 |
1.77 |
15.38 |
19.418 |
3.483 |
T P |
| 2he2_A |
102 |
308 |
67 |
1.94 |
22.39 |
19.385 |
3.279 |
T P |
| 2ozf_A |
92 |
308 |
68 |
2.03 |
20.59 |
19.355 |
3.199 |
T P |
| 2w4f_A |
95 |
308 |
70 |
2.02 |
18.57 |
19.134 |
3.300 |
T P |
| 2he4_A |
90 |
308 |
67 |
1.92 |
16.42 |
19.133 |
3.321 |
T P |
| 1um7_A |
113 |
308 |
66 |
1.86 |
24.24 |
19.117 |
3.361 |
T P |
| 2fcf_A |
89 |
308 |
65 |
1.81 |
20.00 |
19.010 |
3.396 |
T P |
| 2dls_A |
93 |
308 |
64 |
1.80 |
20.31 |
18.871 |
3.366 |
T P |
| 2pkt_A |
91 |
308 |
67 |
1.88 |
17.91 |
18.825 |
3.376 |
T P |
| 2iwq_A |
92 |
308 |
65 |
1.88 |
18.46 |
18.401 |
3.279 |
T P |
| 2vsp_A |
83 |
308 |
65 |
1.90 |
18.46 |
18.192 |
3.256 |
T P |
| 2i6v_A |
87 |
308 |
67 |
1.88 |
17.91 |
17.998 |
3.390 |
T P |
| 2z17_A |
94 |
308 |
64 |
1.70 |
21.88 |
17.945 |
3.556 |
T P |
| 2yuy_A |
126 |
308 |
70 |
2.33 |
15.71 |
17.879 |
2.881 |
T P |
| 2edz_A |
114 |
308 |
68 |
2.10 |
19.12 |
17.814 |
3.095 |
T P |
| 2omj_A |
89 |
308 |
60 |
1.79 |
18.33 |
16.696 |
3.181 |
T P |
| 2i4s_A |
105 |
308 |
67 |
1.86 |
17.91 |
16.356 |
3.413 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]