LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_352.5wLII_11276_180
Total number of 3D structures: 46
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
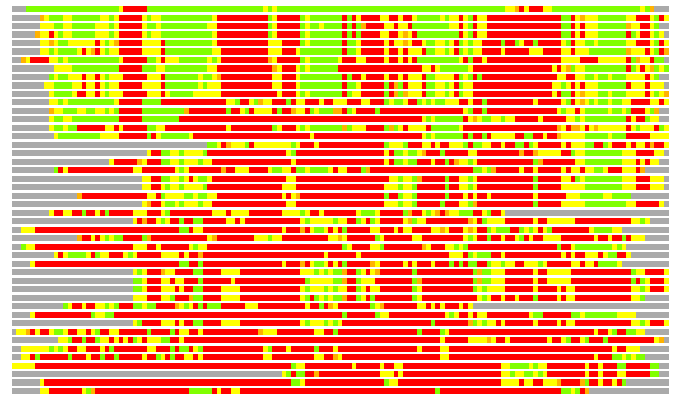
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ft5_A |
211 |
141 |
126 |
1.15 |
26.98 |
88.531 |
10.101 |
T P |
| 2vr0_B |
498 |
141 |
81 |
2.06 |
14.81 |
43.531 |
3.743 |
T P |
| 2j7a_B |
498 |
141 |
82 |
2.14 |
14.63 |
43.404 |
3.660 |
T P |
| 1oah_B |
482 |
141 |
81 |
2.13 |
14.81 |
42.902 |
3.638 |
T P |
| 2rdz_A |
441 |
141 |
85 |
2.40 |
18.82 |
41.091 |
3.404 |
T P |
| 2rf7_A |
441 |
141 |
87 |
2.52 |
17.24 |
39.629 |
3.323 |
T P |
| 3f29_B |
520 |
141 |
77 |
2.25 |
22.08 |
38.942 |
3.282 |
T P |
| 1fgj_A |
499 |
141 |
73 |
2.15 |
26.03 |
37.777 |
3.245 |
T P |
| 1qdb_A |
473 |
141 |
77 |
2.42 |
16.88 |
37.152 |
3.054 |
T P |
| 1fs9_A |
471 |
141 |
80 |
2.44 |
15.00 |
37.116 |
3.153 |
T P |
| 1fs7_A |
471 |
141 |
80 |
2.49 |
15.00 |
37.005 |
3.083 |
T P |
| 2ot4_A |
520 |
141 |
70 |
2.27 |
18.57 |
34.086 |
2.958 |
T P |
| 1sp3_A |
436 |
141 |
67 |
2.34 |
16.42 |
33.215 |
2.745 |
T P |
| 2j7a_C |
145 |
141 |
60 |
2.07 |
20.00 |
33.077 |
2.769 |
T P |
| 3bnj_A |
471 |
141 |
66 |
2.67 |
18.18 |
32.721 |
2.386 |
T P |
| 2ozy_A |
142 |
141 |
62 |
2.25 |
24.19 |
30.837 |
2.643 |
T P |
| 2i5n_C |
332 |
141 |
53 |
2.76 |
3.77 |
22.922 |
1.854 |
T P |
| 1z1n_X |
516 |
141 |
47 |
2.65 |
8.51 |
22.834 |
1.707 |
T P |
| 2e84_A |
513 |
141 |
46 |
2.54 |
17.39 |
22.515 |
1.740 |
T P |
| 2cvc_A |
500 |
141 |
47 |
2.84 |
6.38 |
21.999 |
1.598 |
T P |
| 1duw_A |
289 |
141 |
45 |
2.45 |
8.89 |
21.858 |
1.767 |
T P |
| 19hc_A |
292 |
141 |
47 |
2.51 |
10.64 |
21.845 |
1.798 |
T P |
| 1ofw_A |
292 |
141 |
42 |
2.45 |
7.14 |
21.515 |
1.644 |
T P |
| 1h29_A |
501 |
141 |
45 |
2.53 |
11.11 |
21.333 |
1.713 |
T P |
| 1eys_C |
310 |
141 |
45 |
2.75 |
8.89 |
20.597 |
1.581 |
T P |
| 1kss_A |
568 |
141 |
48 |
2.81 |
4.17 |
20.359 |
1.649 |
T P |
| 2jbl_C |
332 |
141 |
45 |
2.85 |
8.89 |
20.274 |
1.525 |
T P |
| 2b7s_A |
568 |
141 |
43 |
2.79 |
4.65 |
19.455 |
1.490 |
T P |
| 1d4d_A |
560 |
141 |
36 |
2.33 |
0.00 |
19.406 |
1.484 |
T P |
| 1h32_A |
261 |
141 |
39 |
2.83 |
10.26 |
19.404 |
1.329 |
T P |
| 1txw_C |
332 |
141 |
41 |
2.63 |
4.88 |
19.334 |
1.499 |
T P |
| 2b7r_A |
568 |
141 |
44 |
2.76 |
2.27 |
19.244 |
1.540 |
T P |
| 1lj1_A |
568 |
141 |
41 |
2.72 |
2.44 |
19.179 |
1.452 |
T P |
| 1y0p_A |
568 |
141 |
45 |
2.74 |
2.22 |
19.142 |
1.582 |
T P |
| 1jry_A |
568 |
141 |
39 |
2.58 |
5.13 |
18.858 |
1.455 |
T P |
| 2dtg_E |
807 |
141 |
37 |
2.54 |
8.11 |
18.058 |
1.402 |
T P |
| 1m64_A |
568 |
141 |
33 |
2.48 |
12.12 |
17.381 |
1.281 |
T P |
| 1ksu_A |
568 |
141 |
38 |
2.67 |
5.26 |
17.104 |
1.370 |
T P |
| 1e39_A |
568 |
141 |
36 |
2.68 |
5.56 |
16.751 |
1.294 |
T P |
| 1d4c_A |
570 |
141 |
33 |
2.71 |
6.06 |
16.190 |
1.173 |
T P |
| 1q9i_A |
568 |
141 |
33 |
2.83 |
3.03 |
15.832 |
1.127 |
T P |
| 1jrx_A |
568 |
141 |
30 |
2.64 |
3.33 |
15.207 |
1.096 |
T P |
| 1jrz_A |
568 |
141 |
28 |
2.68 |
0.00 |
14.556 |
1.009 |
T P |
| 1qo8_A |
564 |
141 |
25 |
2.57 |
0.00 |
13.863 |
0.936 |
T P |
| 1p2e_A |
568 |
141 |
22 |
2.82 |
0.00 |
11.660 |
0.754 |
T P |
| 1p2h_A |
568 |
141 |
19 |
2.34 |
5.26 |
10.134 |
0.778 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]