LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_356.5wLII_11277_3
Total number of 3D structures: 53
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
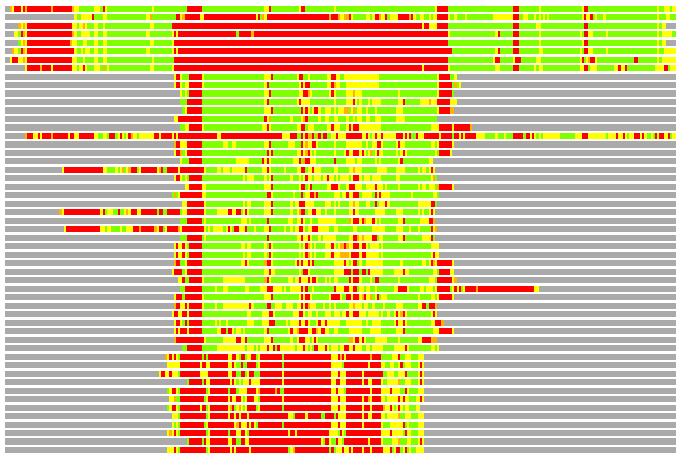
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1zfj_A |
476 |
310 |
263 |
1.32 |
12.93 |
82.189 |
18.508 |
T P |
| 1jr1_A |
436 |
310 |
191 |
2.04 |
13.61 |
48.751 |
8.915 |
T P |
| 1vrd_A |
321 |
310 |
151 |
1.53 |
13.25 |
46.621 |
9.270 |
T P |
| 1me7_A |
365 |
310 |
154 |
1.68 |
11.69 |
46.154 |
8.633 |
T P |
| 1eep_A |
314 |
310 |
148 |
1.48 |
12.84 |
45.880 |
9.383 |
T P |
| 1me8_A |
357 |
310 |
150 |
1.67 |
12.00 |
45.097 |
8.487 |
T P |
| 2cu0_A |
358 |
310 |
147 |
1.59 |
10.20 |
44.696 |
8.714 |
T P |
| 1jcn_A |
395 |
310 |
146 |
1.83 |
13.01 |
41.289 |
7.566 |
T P |
| 2qrd_G |
325 |
310 |
113 |
1.67 |
11.50 |
33.981 |
6.373 |
T P |
| 2oox_E |
333 |
310 |
114 |
1.77 |
13.16 |
33.858 |
6.096 |
T P |
| 2v8q_E |
304 |
310 |
111 |
1.60 |
18.92 |
33.771 |
6.548 |
T P |
| 2o16_B |
135 |
310 |
111 |
1.79 |
17.12 |
32.923 |
5.867 |
T P |
| 3ddj_A |
279 |
310 |
109 |
1.88 |
21.10 |
32.265 |
5.494 |
T P |
| 2rih_A |
131 |
310 |
109 |
1.84 |
16.51 |
31.983 |
5.615 |
T P |
| 2qlv_C |
310 |
310 |
108 |
1.81 |
14.81 |
31.933 |
5.667 |
T P |
| 1nf7_A |
454 |
310 |
151 |
2.70 |
9.27 |
31.785 |
5.398 |
T P |
| 2p9m_B |
129 |
310 |
108 |
1.88 |
14.81 |
31.591 |
5.450 |
T P |
| 2rc3_C |
128 |
310 |
104 |
1.80 |
15.38 |
30.855 |
5.483 |
T P |
| 3fna_B |
123 |
310 |
104 |
1.78 |
15.38 |
30.833 |
5.540 |
T P |
| 2oux_A |
257 |
310 |
122 |
2.24 |
17.21 |
30.613 |
5.211 |
T P |
| 2uv7_A |
143 |
310 |
108 |
2.00 |
16.67 |
30.392 |
5.134 |
T P |
| 2nyc_A |
130 |
310 |
102 |
1.88 |
12.75 |
29.594 |
5.148 |
T P |
| 1o50_A |
141 |
310 |
102 |
1.84 |
15.69 |
29.529 |
5.270 |
T P |
| 1vr9_A |
121 |
310 |
102 |
1.99 |
18.63 |
28.824 |
4.871 |
T P |
| 2yvx_A |
442 |
310 |
117 |
2.18 |
18.80 |
28.725 |
5.128 |
T P |
| 1xkf_B |
123 |
310 |
101 |
1.97 |
15.84 |
28.639 |
4.871 |
T P |
| 2yvy_A |
248 |
310 |
120 |
2.31 |
17.50 |
28.220 |
4.973 |
T P |
| 1y5h_A |
123 |
310 |
102 |
1.88 |
15.69 |
26.976 |
5.161 |
T P |
| 2uv4_A |
143 |
310 |
106 |
2.00 |
19.81 |
26.830 |
5.046 |
T P |
| 2uv6_A |
143 |
310 |
105 |
2.04 |
20.00 |
26.574 |
4.917 |
T P |
| 2yzq_A |
224 |
310 |
103 |
2.05 |
22.33 |
26.504 |
4.781 |
T P |
| 3fhm_A |
136 |
310 |
102 |
1.92 |
18.63 |
26.172 |
5.038 |
T P |
| 3ctu_B |
152 |
310 |
104 |
2.05 |
18.27 |
26.155 |
4.840 |
T P |
| 2yzi_B |
136 |
310 |
105 |
2.13 |
15.24 |
25.948 |
4.706 |
T P |
| 2nye_A |
132 |
310 |
98 |
1.73 |
12.24 |
25.874 |
5.343 |
T P |
| 2ef7_A |
127 |
310 |
103 |
2.31 |
13.59 |
25.491 |
4.267 |
T P |
| 2qh1_B |
185 |
310 |
102 |
2.14 |
13.73 |
24.717 |
4.544 |
T P |
| 1pvm_B |
184 |
310 |
104 |
2.21 |
16.35 |
24.479 |
4.506 |
T P |
| 2j9l_C |
172 |
310 |
99 |
2.10 |
20.20 |
24.210 |
4.497 |
T P |
| 2yvz_A |
248 |
310 |
94 |
2.26 |
20.21 |
24.167 |
3.991 |
T P |
| 1pbj_A |
120 |
310 |
101 |
2.38 |
14.85 |
23.184 |
4.080 |
T P |
| 2r2z_A |
84 |
310 |
42 |
2.50 |
9.52 |
9.809 |
1.614 |
T P |
| 3ded_C |
91 |
310 |
44 |
2.54 |
4.55 |
9.639 |
1.667 |
T P |
| 2pli_A |
84 |
310 |
45 |
2.67 |
8.89 |
9.374 |
1.627 |
T P |
| 2o1r_A |
81 |
310 |
44 |
2.73 |
4.55 |
9.078 |
1.552 |
T P |
| 2nqw_A |
87 |
310 |
42 |
2.75 |
4.76 |
9.065 |
1.472 |
T P |
| 2p13_A |
85 |
310 |
43 |
2.84 |
4.65 |
9.021 |
1.465 |
T P |
| 2pls_A |
86 |
310 |
41 |
2.82 |
4.88 |
8.966 |
1.404 |
T P |
| 2p4p_A |
84 |
310 |
44 |
2.74 |
6.82 |
8.872 |
1.550 |
T P |
| 2o3g_A |
76 |
310 |
41 |
2.66 |
7.32 |
8.786 |
1.483 |
T P |
| 2rk5_A |
86 |
310 |
39 |
2.66 |
5.13 |
8.560 |
1.416 |
T P |
| 2oai_A |
80 |
310 |
38 |
2.47 |
5.26 |
8.332 |
1.476 |
T P |
| 2p3h_A |
98 |
310 |
38 |
2.71 |
5.26 |
8.048 |
1.353 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]