LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_357.5wLII_11277_4
Total number of 3D structures: 64
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
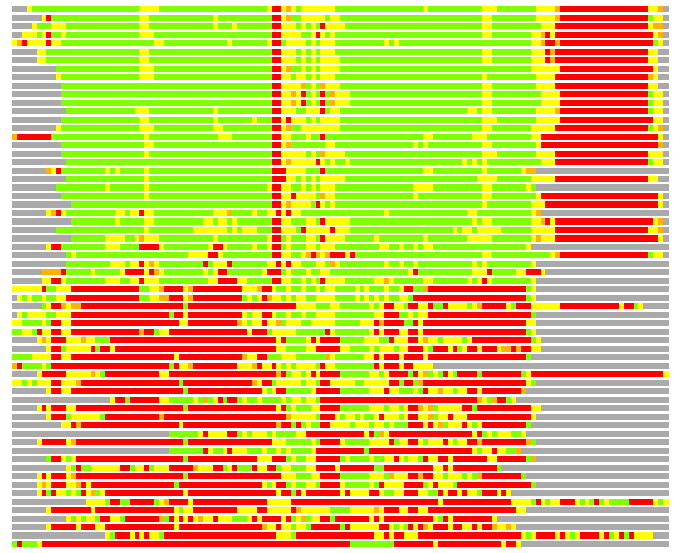
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2vuo_A |
215 |
134 |
110 |
1.80 |
17.27 |
74.669 |
5.794 |
T P |
| 2dtq_A |
211 |
134 |
106 |
1.61 |
14.15 |
73.769 |
6.208 |
T P |
| 1iix_B |
213 |
134 |
107 |
1.77 |
14.02 |
73.620 |
5.728 |
T P |
| 1e4k_A |
216 |
134 |
106 |
1.68 |
14.15 |
73.138 |
5.949 |
T P |
| 1hzh_H |
457 |
134 |
108 |
1.90 |
14.81 |
72.759 |
5.400 |
T P |
| 1t83_B |
212 |
134 |
105 |
1.73 |
14.29 |
72.642 |
5.735 |
T P |
| 1iis_B |
212 |
134 |
105 |
1.74 |
14.29 |
72.520 |
5.697 |
T P |
| 3c2s_A |
210 |
134 |
102 |
1.63 |
13.73 |
72.051 |
5.893 |
T P |
| 2j6e_A |
209 |
134 |
102 |
1.61 |
13.73 |
71.364 |
5.968 |
T P |
| 1l6x_A |
207 |
134 |
101 |
1.61 |
13.86 |
71.350 |
5.895 |
T P |
| 2gj7_A |
207 |
134 |
101 |
1.53 |
13.86 |
70.933 |
6.179 |
T P |
| 1dn2_A |
207 |
134 |
101 |
1.53 |
13.86 |
70.933 |
6.180 |
T P |
| 1h3t_A |
207 |
134 |
101 |
1.59 |
13.86 |
70.547 |
5.989 |
T P |
| 2wah_B |
209 |
134 |
101 |
1.67 |
13.86 |
70.450 |
5.694 |
T P |
| 2ql1_A |
209 |
134 |
103 |
1.71 |
13.59 |
70.190 |
5.699 |
T P |
| 1igt_B |
444 |
134 |
99 |
1.53 |
15.15 |
70.078 |
6.084 |
T P |
| 2rgs_B |
207 |
134 |
97 |
1.47 |
14.43 |
69.576 |
6.192 |
T P |
| 1oqo_A |
208 |
134 |
102 |
1.69 |
13.73 |
69.414 |
5.699 |
T P |
| 1fc2_D |
206 |
134 |
100 |
1.66 |
14.00 |
68.986 |
5.670 |
T P |
| 1mco_H |
428 |
134 |
95 |
1.54 |
13.68 |
68.047 |
5.775 |
T P |
| 1fcc_A |
206 |
134 |
99 |
1.77 |
14.14 |
67.694 |
5.283 |
T P |
| 3dj9_A |
107 |
134 |
96 |
1.44 |
13.54 |
67.416 |
6.223 |
T P |
| 2vol_A |
207 |
134 |
96 |
1.57 |
12.50 |
67.198 |
5.762 |
T P |
| 1frt_C |
205 |
134 |
95 |
1.56 |
14.74 |
67.115 |
5.730 |
T P |
| 1ls0_B |
320 |
134 |
97 |
1.83 |
21.65 |
67.032 |
5.031 |
T P |
| 1i1a_C |
205 |
134 |
97 |
1.82 |
14.43 |
66.062 |
5.052 |
T P |
| 1igy_B |
434 |
134 |
101 |
2.04 |
12.87 |
65.996 |
4.729 |
T P |
| 1i1c_A |
205 |
134 |
97 |
1.98 |
13.40 |
64.175 |
4.661 |
T P |
| 1ow0_A |
209 |
134 |
89 |
1.66 |
14.61 |
61.336 |
5.067 |
T P |
| 1adq_A |
206 |
134 |
95 |
1.69 |
13.68 |
58.397 |
5.313 |
T P |
| 1fp5_A |
208 |
134 |
91 |
1.83 |
21.98 |
57.714 |
4.723 |
T P |
| 2ig2_H |
238 |
134 |
87 |
1.99 |
12.64 |
57.191 |
4.156 |
T P |
| 2qej_A |
211 |
134 |
90 |
1.98 |
8.89 |
51.002 |
4.327 |
T P |
| 2dsb_A |
206 |
134 |
54 |
2.42 |
3.70 |
28.589 |
2.145 |
T P |
| 2dsc_A |
195 |
134 |
54 |
2.44 |
3.70 |
28.273 |
2.123 |
T P |
| 1ihu_A |
540 |
134 |
50 |
2.60 |
8.00 |
26.031 |
1.851 |
T P |
| 3bm4_A |
197 |
134 |
47 |
2.43 |
6.38 |
25.548 |
1.859 |
T P |
| 1viq_A |
204 |
134 |
45 |
2.51 |
8.89 |
24.110 |
1.723 |
T P |
| 1mk1_A |
187 |
134 |
41 |
2.45 |
2.44 |
23.306 |
1.608 |
T P |
| 1g20_H |
262 |
134 |
45 |
2.79 |
4.44 |
22.778 |
1.558 |
T P |
| 2ved_B |
254 |
134 |
43 |
2.83 |
9.30 |
22.542 |
1.467 |
T P |
| 1vhz_A |
186 |
134 |
40 |
2.45 |
2.50 |
22.297 |
1.568 |
T P |
| 1viu_B |
181 |
134 |
42 |
2.58 |
0.00 |
21.923 |
1.565 |
T P |
| 3end_A |
268 |
134 |
40 |
2.58 |
7.50 |
21.689 |
1.494 |
T P |
| 1vhg_A |
185 |
134 |
42 |
2.58 |
2.38 |
21.438 |
1.570 |
T P |
| 1wcv_1 |
243 |
134 |
39 |
2.51 |
5.13 |
21.298 |
1.497 |
T P |
| 2oze_A |
284 |
134 |
38 |
2.41 |
7.89 |
20.941 |
1.512 |
T P |
| 2afh_E |
289 |
134 |
39 |
2.49 |
12.82 |
20.936 |
1.505 |
T P |
| 2ph1_A |
247 |
134 |
41 |
2.90 |
7.32 |
20.934 |
1.369 |
T P |
| 1xd8_A |
289 |
134 |
37 |
2.76 |
10.81 |
20.906 |
1.296 |
T P |
| 2yvu_A |
179 |
134 |
39 |
2.28 |
2.56 |
20.740 |
1.641 |
T P |
| 3bfv_A |
241 |
134 |
39 |
2.48 |
10.26 |
20.721 |
1.510 |
T P |
| 3cwq_A |
206 |
134 |
38 |
2.20 |
5.26 |
20.675 |
1.655 |
T P |
| 2bek_A |
244 |
134 |
37 |
2.42 |
5.41 |
20.550 |
1.466 |
T P |
| 1rw4_A |
271 |
134 |
43 |
2.61 |
11.63 |
20.524 |
1.586 |
T P |
| 2c8v_A |
271 |
134 |
39 |
2.61 |
10.26 |
20.503 |
1.438 |
T P |
| 1g3q_A |
237 |
134 |
37 |
2.57 |
2.70 |
20.423 |
1.388 |
T P |
| 1xdb_A |
289 |
134 |
36 |
2.80 |
2.78 |
20.053 |
1.240 |
T P |
| 2dpo_A |
310 |
134 |
39 |
2.69 |
5.13 |
19.871 |
1.400 |
T P |
| 1g0s_B |
202 |
134 |
39 |
2.91 |
7.69 |
19.620 |
1.294 |
T P |
| 1de0_A |
289 |
134 |
40 |
2.74 |
0.00 |
19.115 |
1.406 |
T P |
| 1ion_A |
243 |
134 |
37 |
3.08 |
5.41 |
18.326 |
1.165 |
T P |
| 1hyq_A |
232 |
134 |
32 |
2.68 |
9.38 |
15.792 |
1.149 |
T P |
| 2yvp_A |
182 |
134 |
17 |
2.07 |
0.00 |
10.472 |
0.784 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]