LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_364.5wLII_11277_25
Total number of 3D structures: 37
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
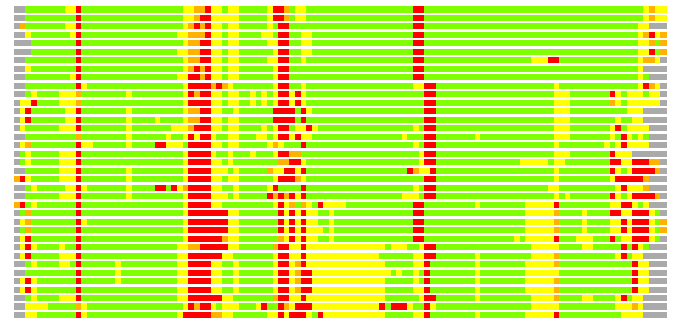
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1a8s_A |
273 |
116 |
108 |
1.57 |
16.67 |
89.780 |
6.449 |
T P |
| 1va4_A |
271 |
116 |
107 |
1.50 |
15.89 |
89.682 |
6.690 |
T P |
| 1a8q_A |
274 |
116 |
105 |
1.25 |
13.33 |
88.449 |
7.797 |
T P |
| 1hkh_A |
279 |
116 |
107 |
1.58 |
16.82 |
88.328 |
6.370 |
T P |
| 1bro_A |
277 |
116 |
106 |
1.58 |
14.15 |
88.279 |
6.292 |
T P |
| 1a8u_A |
277 |
116 |
105 |
1.48 |
14.29 |
87.995 |
6.638 |
T P |
| 1a88_A |
275 |
116 |
104 |
1.53 |
14.42 |
87.020 |
6.396 |
T P |
| 3fob_A |
277 |
116 |
103 |
1.31 |
13.59 |
86.664 |
7.309 |
T P |
| 1brt_A |
277 |
116 |
103 |
1.20 |
14.56 |
86.479 |
7.895 |
T P |
| 1zoi_A |
275 |
116 |
102 |
1.49 |
13.73 |
86.092 |
6.418 |
T P |
| 2e3j_A |
346 |
116 |
104 |
1.76 |
9.62 |
82.877 |
5.593 |
T P |
| 2zjf_A |
346 |
116 |
104 |
1.78 |
9.62 |
82.707 |
5.518 |
T P |
| 2d0d_A |
271 |
116 |
100 |
1.64 |
13.00 |
81.719 |
5.753 |
T P |
| 1iup_A |
271 |
116 |
100 |
1.66 |
12.00 |
81.605 |
5.684 |
T P |
| 2vf2_A |
284 |
116 |
102 |
1.79 |
8.82 |
81.058 |
5.392 |
T P |
| 1y37_A |
294 |
116 |
102 |
1.74 |
13.73 |
80.939 |
5.542 |
T P |
| 1wm1_A |
313 |
116 |
101 |
1.82 |
10.89 |
78.567 |
5.262 |
T P |
| 1zd3_A |
547 |
116 |
99 |
1.74 |
8.08 |
76.788 |
5.385 |
T P |
| 1cqz_B |
541 |
116 |
100 |
1.88 |
8.00 |
76.595 |
5.063 |
T P |
| 3cxu_A |
320 |
116 |
98 |
1.74 |
12.24 |
76.339 |
5.326 |
T P |
| 1ehy_A |
282 |
116 |
97 |
1.65 |
16.49 |
76.176 |
5.534 |
T P |
| 1azw_A |
313 |
116 |
98 |
1.67 |
11.22 |
75.877 |
5.543 |
T P |
| 2cjp_A |
320 |
116 |
97 |
1.73 |
13.40 |
75.519 |
5.289 |
T P |
| 1hde_A |
310 |
116 |
100 |
1.85 |
11.00 |
72.415 |
5.139 |
T P |
| 1b6g_A |
308 |
116 |
96 |
1.77 |
10.42 |
69.093 |
5.133 |
T P |
| 1bez_A |
310 |
116 |
98 |
1.86 |
10.20 |
69.029 |
4.994 |
T P |
| 2yxp_X |
310 |
116 |
98 |
1.85 |
10.20 |
68.976 |
5.024 |
T P |
| 1be0_A |
310 |
116 |
97 |
1.79 |
10.31 |
68.855 |
5.128 |
T P |
| 2v9z_A |
302 |
116 |
98 |
2.02 |
13.27 |
67.029 |
4.616 |
T P |
| 1cqw_A |
295 |
116 |
97 |
1.96 |
13.40 |
66.760 |
4.709 |
T P |
| 1mj5_A |
297 |
116 |
101 |
2.00 |
12.87 |
66.008 |
4.812 |
T P |
| 1iz7_A |
294 |
116 |
100 |
1.97 |
13.00 |
65.333 |
4.839 |
T P |
| 1cv2_A |
293 |
116 |
100 |
1.99 |
13.00 |
65.169 |
4.795 |
T P |
| 1g42_A |
293 |
116 |
100 |
2.01 |
13.00 |
65.124 |
4.750 |
T P |
| 1bn6_A |
291 |
116 |
97 |
1.99 |
13.40 |
64.740 |
4.639 |
T P |
| 2o2i_A |
293 |
116 |
97 |
2.14 |
13.40 |
62.218 |
4.324 |
T P |
| 2qvb_A |
296 |
116 |
97 |
2.11 |
13.40 |
61.917 |
4.386 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]