LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_372.5wLII_11277_56
Total number of 3D structures: 47
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
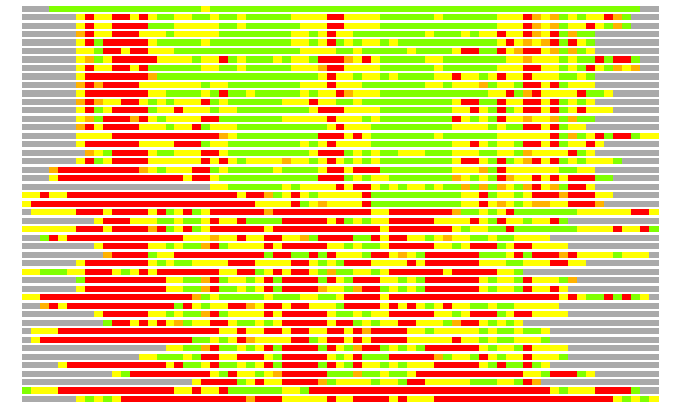
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1rws_A |
68 |
71 |
66 |
0.70 |
22.73 |
91.654 |
8.287 |
T P |
| 1fm0_D |
81 |
71 |
54 |
2.39 |
16.67 |
57.962 |
2.169 |
T P |
| 3bii_D |
80 |
71 |
53 |
2.34 |
13.21 |
57.234 |
2.169 |
T P |
| 1ryj_A |
70 |
71 |
50 |
2.25 |
26.00 |
56.729 |
2.128 |
T P |
| 1jsb_A |
70 |
71 |
48 |
2.07 |
25.00 |
56.081 |
2.213 |
T P |
| 1xo3_A |
101 |
71 |
49 |
2.28 |
14.29 |
55.918 |
2.061 |
T P |
| 2q5w_D |
77 |
71 |
50 |
2.29 |
20.00 |
53.354 |
2.091 |
T P |
| 1nvi_D |
81 |
71 |
54 |
2.44 |
16.67 |
52.895 |
2.126 |
T P |
| 2cu3_B |
64 |
71 |
47 |
2.15 |
14.89 |
51.599 |
2.093 |
T P |
| 1zud_4 |
66 |
71 |
49 |
2.42 |
12.24 |
51.398 |
1.943 |
T P |
| 1nyr_A |
642 |
71 |
48 |
2.30 |
10.42 |
51.162 |
2.004 |
T P |
| 1wgk_A |
114 |
71 |
47 |
2.29 |
10.64 |
50.371 |
1.968 |
T P |
| 1vjk_A |
87 |
71 |
45 |
2.25 |
11.11 |
50.122 |
1.913 |
T P |
| 2qjl_A |
99 |
71 |
49 |
2.50 |
8.16 |
50.007 |
1.887 |
T P |
| 1tyg_B |
65 |
71 |
47 |
2.30 |
14.89 |
49.650 |
1.959 |
T P |
| 1v8c_A |
165 |
71 |
45 |
2.25 |
24.44 |
49.376 |
1.911 |
T P |
| 3cwi_A |
69 |
71 |
44 |
2.39 |
22.73 |
47.508 |
1.768 |
T P |
| 2g1e_A |
90 |
71 |
47 |
2.35 |
19.15 |
46.805 |
1.917 |
T P |
| 3dwg_C |
92 |
71 |
48 |
2.55 |
8.33 |
46.358 |
1.809 |
T P |
| 2k5p_A |
78 |
71 |
44 |
2.40 |
20.45 |
43.841 |
1.761 |
T P |
| 2ax5_A |
99 |
71 |
38 |
2.13 |
10.53 |
40.451 |
1.702 |
T P |
| 1fjg_D |
208 |
71 |
37 |
2.45 |
2.70 |
38.477 |
1.454 |
T P |
| 2vqe_D |
208 |
71 |
35 |
2.67 |
2.86 |
35.699 |
1.264 |
T P |
| 1n32_D |
208 |
71 |
34 |
2.83 |
8.82 |
34.845 |
1.160 |
T P |
| 3dva_B |
324 |
71 |
34 |
2.73 |
2.94 |
33.308 |
1.201 |
T P |
| 3fz4_A |
119 |
71 |
34 |
2.46 |
2.94 |
32.817 |
1.329 |
T P |
| 1w85_B |
324 |
71 |
34 |
2.79 |
2.94 |
32.660 |
1.175 |
T P |
| 3f0i_A |
119 |
71 |
33 |
2.77 |
9.09 |
30.108 |
1.149 |
T P |
| 1s3c_A |
138 |
71 |
31 |
2.52 |
9.68 |
30.056 |
1.185 |
T P |
| 1j9b_A |
137 |
71 |
30 |
2.73 |
3.33 |
29.978 |
1.061 |
T P |
| 1sk1_A |
137 |
71 |
33 |
2.83 |
9.09 |
29.759 |
1.128 |
T P |
| 2hj1_B |
80 |
71 |
29 |
2.44 |
0.00 |
29.452 |
1.142 |
T P |
| 1sk0_A |
137 |
71 |
30 |
2.52 |
10.00 |
29.215 |
1.146 |
T P |
| 1sd8_A |
138 |
71 |
30 |
2.63 |
13.33 |
29.122 |
1.098 |
T P |
| 3b98_A |
446 |
71 |
27 |
2.33 |
7.41 |
29.023 |
1.112 |
T P |
| 1rw1_A |
114 |
71 |
31 |
2.56 |
9.68 |
28.976 |
1.164 |
T P |
| 1sk2_A |
138 |
71 |
31 |
2.67 |
9.68 |
28.927 |
1.120 |
T P |
| 1wik_A |
109 |
71 |
30 |
2.60 |
6.67 |
28.923 |
1.110 |
T P |
| 3gfk_A |
118 |
71 |
28 |
2.74 |
3.57 |
28.868 |
0.986 |
T P |
| 1z3e_A |
119 |
71 |
30 |
2.62 |
6.67 |
28.497 |
1.101 |
T P |
| 1i9d_A |
138 |
71 |
29 |
2.52 |
10.34 |
28.237 |
1.107 |
T P |
| 1jzw_A |
137 |
71 |
30 |
2.63 |
16.67 |
27.826 |
1.099 |
T P |
| 1s3d_A |
138 |
71 |
28 |
2.46 |
10.71 |
27.612 |
1.094 |
T P |
| 1fov_A |
82 |
71 |
24 |
2.41 |
16.67 |
25.378 |
0.955 |
T P |
| 3grx_A |
82 |
71 |
27 |
2.63 |
7.41 |
25.366 |
0.989 |
T P |
| 1r7h_A |
74 |
71 |
23 |
2.31 |
4.35 |
25.351 |
0.955 |
T P |
| 1nm3_B |
239 |
71 |
22 |
2.79 |
9.09 |
21.832 |
0.761 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]