LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_375.5wLII_11277_59
Total number of 3D structures: 76
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
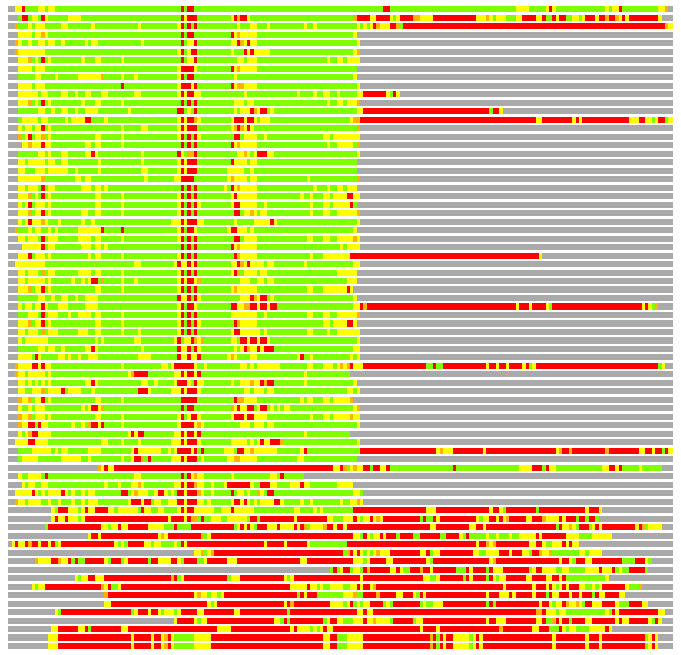
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ys7_B |
226 |
200 |
189 |
1.39 |
13.76 |
89.651 |
12.652 |
T P |
| 2gwr_A |
225 |
200 |
134 |
2.04 |
14.93 |
55.251 |
6.251 |
T P |
| 1w25_A |
454 |
200 |
113 |
1.80 |
10.62 |
51.638 |
5.934 |
T P |
| 2eub_A |
121 |
200 |
98 |
1.59 |
12.24 |
46.673 |
5.794 |
T P |
| 1zes_A |
121 |
200 |
100 |
1.67 |
14.00 |
46.211 |
5.657 |
T P |
| 1mvo_A |
121 |
200 |
99 |
1.68 |
12.12 |
46.158 |
5.562 |
T P |
| 1e6k_A |
130 |
200 |
100 |
1.79 |
13.00 |
46.130 |
5.299 |
T P |
| 2a9o_A |
117 |
200 |
97 |
1.52 |
14.43 |
46.050 |
5.981 |
T P |
| 1dbw_B |
125 |
200 |
99 |
1.57 |
8.08 |
46.018 |
5.922 |
T P |
| 2a9r_A |
117 |
200 |
97 |
1.59 |
13.40 |
45.943 |
5.730 |
T P |
| 1ny5_B |
385 |
200 |
104 |
1.90 |
8.65 |
45.817 |
5.212 |
T P |
| 1ymv_A |
124 |
200 |
99 |
1.73 |
13.13 |
45.789 |
5.419 |
T P |
| 2jba_A |
125 |
200 |
100 |
1.70 |
13.00 |
45.656 |
5.567 |
T P |
| 1p2f_A |
217 |
200 |
106 |
1.96 |
8.49 |
45.248 |
5.137 |
T P |
| 1zh2_A |
120 |
200 |
96 |
1.57 |
14.58 |
45.189 |
5.736 |
T P |
| 1ymu_A |
125 |
200 |
97 |
1.79 |
11.34 |
45.132 |
5.130 |
T P |
| 2j8e_A |
128 |
200 |
97 |
1.81 |
13.40 |
44.947 |
5.083 |
T P |
| 2iyn_B |
124 |
200 |
97 |
1.73 |
13.40 |
44.939 |
5.295 |
T P |
| 1xhf_B |
122 |
200 |
97 |
1.64 |
9.28 |
44.829 |
5.590 |
T P |
| 1xhe_B |
122 |
200 |
98 |
1.67 |
10.20 |
44.759 |
5.538 |
T P |
| 1zgz_A |
121 |
200 |
99 |
1.77 |
12.12 |
44.730 |
5.280 |
T P |
| 1kmi_Y |
128 |
200 |
97 |
1.81 |
11.34 |
44.536 |
5.076 |
T P |
| 1d4z_A |
128 |
200 |
96 |
1.87 |
13.54 |
44.526 |
4.864 |
T P |
| 1ab5_A |
125 |
200 |
95 |
1.75 |
11.58 |
44.510 |
5.148 |
T P |
| 1udr_A |
126 |
200 |
97 |
1.91 |
12.37 |
44.478 |
4.823 |
T P |
| 2rjn_A |
135 |
200 |
97 |
1.78 |
13.40 |
44.461 |
5.156 |
T P |
| 1d5w_A |
122 |
200 |
96 |
1.69 |
8.33 |
44.432 |
5.372 |
T P |
| 3chy_A |
128 |
200 |
97 |
1.91 |
10.31 |
44.419 |
4.826 |
T P |
| 1mih_A |
128 |
200 |
97 |
1.87 |
12.37 |
44.409 |
4.922 |
T P |
| 2che_A |
128 |
200 |
96 |
1.88 |
14.58 |
44.317 |
4.856 |
T P |
| 2zay_A |
123 |
200 |
99 |
1.84 |
10.10 |
44.296 |
5.095 |
T P |
| 2fka_A |
128 |
200 |
98 |
1.88 |
12.24 |
44.289 |
4.947 |
T P |
| 1qkk_A |
139 |
200 |
97 |
1.81 |
10.31 |
44.110 |
5.078 |
T P |
| 1ehc_A |
128 |
200 |
96 |
1.89 |
13.54 |
44.077 |
4.834 |
T P |
| 2jba_B |
121 |
200 |
96 |
1.62 |
13.54 |
44.070 |
5.583 |
T P |
| 1kgs_A |
219 |
200 |
100 |
1.94 |
11.00 |
44.058 |
4.900 |
T P |
| 1hey_A |
127 |
200 |
99 |
1.96 |
11.11 |
44.024 |
4.814 |
T P |
| 1ab6_A |
125 |
200 |
95 |
1.86 |
13.68 |
43.950 |
4.847 |
T P |
| 1e6l_A |
127 |
200 |
98 |
1.95 |
10.20 |
43.921 |
4.782 |
T P |
| 3cg4_A |
126 |
200 |
95 |
1.81 |
6.32 |
43.833 |
4.976 |
T P |
| 2jb9_B |
122 |
200 |
94 |
1.68 |
13.83 |
43.818 |
5.287 |
T P |
| 3bre_B |
328 |
200 |
98 |
1.89 |
8.16 |
43.646 |
4.926 |
T P |
| 1s8n_A |
190 |
200 |
107 |
2.12 |
12.15 |
43.468 |
4.814 |
T P |
| 1tmy_A |
118 |
200 |
93 |
1.64 |
8.60 |
43.401 |
5.333 |
T P |
| 1b00_A |
122 |
200 |
94 |
1.74 |
12.77 |
43.255 |
5.106 |
T P |
| 2qr3_A |
121 |
200 |
95 |
1.78 |
9.47 |
43.250 |
5.062 |
T P |
| 1jbe_A |
126 |
200 |
94 |
1.93 |
13.83 |
43.026 |
4.633 |
T P |
| 1yio_A |
198 |
200 |
93 |
1.77 |
11.83 |
42.890 |
4.971 |
T P |
| 1cye_A |
129 |
200 |
95 |
1.92 |
12.63 |
42.882 |
4.693 |
T P |
| 1l5z_A |
146 |
200 |
93 |
1.81 |
9.68 |
42.794 |
4.870 |
T P |
| 1u0s_Y |
118 |
200 |
91 |
1.61 |
7.69 |
42.537 |
5.309 |
T P |
| 3cu5_B |
129 |
200 |
94 |
1.90 |
10.64 |
41.591 |
4.695 |
T P |
| 1a2o_A |
347 |
200 |
108 |
2.24 |
9.26 |
40.950 |
4.607 |
T P |
| 2qxy_A |
119 |
200 |
95 |
2.00 |
11.58 |
39.221 |
4.526 |
T P |
| 2oqr_A |
226 |
200 |
89 |
1.91 |
11.24 |
38.979 |
4.432 |
T P |
| 1dz3_A |
123 |
200 |
82 |
1.64 |
6.10 |
38.573 |
4.722 |
T P |
| 2jvj_A |
132 |
200 |
86 |
2.05 |
13.95 |
33.704 |
4.008 |
T P |
| 1dc8_A |
123 |
200 |
91 |
2.07 |
9.89 |
33.561 |
4.203 |
T P |
| 1dc7_A |
124 |
200 |
90 |
2.22 |
11.11 |
31.141 |
3.886 |
T P |
| 1a04_A |
205 |
200 |
87 |
2.28 |
11.49 |
30.639 |
3.658 |
T P |
| 2vxo_B |
658 |
200 |
66 |
2.62 |
4.55 |
20.967 |
2.425 |
T P |
| 1gyt_A |
503 |
200 |
63 |
2.77 |
6.35 |
19.855 |
2.192 |
T P |
| 2dqb_C |
364 |
200 |
58 |
2.43 |
12.07 |
19.409 |
2.294 |
T P |
| 3ezu_A |
336 |
200 |
53 |
2.59 |
1.89 |
18.582 |
1.974 |
T P |
| 2pq7_A |
174 |
200 |
54 |
2.68 |
9.26 |
18.023 |
1.939 |
T P |
| 1dgj_A |
906 |
200 |
52 |
2.75 |
3.85 |
17.142 |
1.823 |
T P |
| 1xx7_A |
172 |
200 |
50 |
2.47 |
6.00 |
16.957 |
1.943 |
T P |
| 2o08_A |
187 |
200 |
48 |
2.50 |
4.17 |
16.906 |
1.847 |
T P |
| 3ccg_A |
189 |
200 |
50 |
2.75 |
6.00 |
16.380 |
1.755 |
T P |
| 2ogi_B |
194 |
200 |
48 |
2.73 |
4.17 |
16.150 |
1.699 |
T P |
| 1yoy_A |
146 |
200 |
50 |
2.60 |
6.00 |
15.911 |
1.850 |
T P |
| 2o6i_A |
436 |
200 |
46 |
2.50 |
0.00 |
15.893 |
1.773 |
T P |
| 2hek_A |
369 |
200 |
49 |
2.77 |
8.16 |
15.699 |
1.705 |
T P |
| 3dto_A |
189 |
200 |
44 |
2.79 |
6.82 |
15.625 |
1.524 |
T P |
| 1vlb_A |
907 |
200 |
46 |
2.67 |
2.17 |
14.759 |
1.662 |
T P |
| 1hlr_A |
907 |
200 |
46 |
2.67 |
2.17 |
14.759 |
1.662 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]