LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_377.5wLII_11277_67
Total number of 3D structures: 40
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
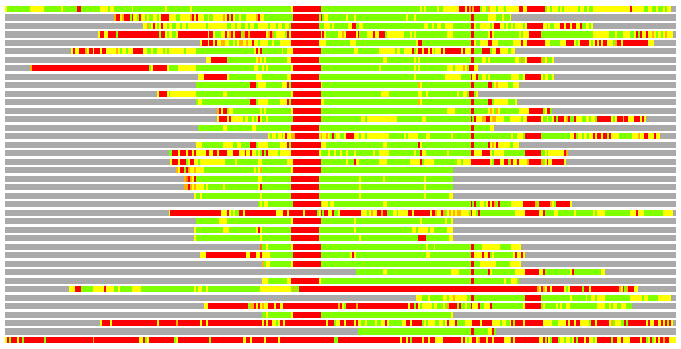
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1w3b_A |
388 |
344 |
306 |
1.88 |
11.11 |
80.592 |
15.423 |
T P |
| 2q7f_A |
194 |
344 |
172 |
1.93 |
5.23 |
44.771 |
8.469 |
T P |
| 2vq2_A |
220 |
344 |
193 |
2.24 |
8.29 |
44.223 |
8.241 |
T P |
| 2gw1_A |
487 |
344 |
194 |
2.19 |
7.73 |
42.249 |
8.461 |
T P |
| 2ho1_A |
222 |
344 |
168 |
2.11 |
11.31 |
41.749 |
7.597 |
T P |
| 2fi7_A |
223 |
344 |
172 |
2.16 |
9.30 |
40.621 |
7.602 |
T P |
| 2c0l_A |
292 |
344 |
148 |
1.54 |
6.08 |
40.481 |
9.005 |
T P |
| 2c0m_C |
302 |
344 |
146 |
1.67 |
8.22 |
39.720 |
8.238 |
T P |
| 1fch_A |
302 |
344 |
147 |
1.70 |
5.44 |
39.401 |
8.178 |
T P |
| 1e96_B |
185 |
344 |
143 |
1.49 |
11.19 |
39.392 |
8.987 |
T P |
| 3cv0_A |
300 |
344 |
144 |
1.67 |
10.42 |
38.987 |
8.123 |
T P |
| 1hh8_A |
192 |
344 |
142 |
1.50 |
11.27 |
38.813 |
8.880 |
T P |
| 3cvq_A |
289 |
344 |
144 |
1.67 |
6.25 |
38.754 |
8.144 |
T P |
| 1xnf_B |
262 |
344 |
167 |
2.17 |
3.59 |
38.477 |
7.353 |
T P |
| 2fo7_A |
136 |
344 |
136 |
1.09 |
15.44 |
38.071 |
11.458 |
T P |
| 2pl2_A |
194 |
344 |
163 |
2.22 |
11.66 |
38.003 |
7.035 |
T P |
| 1wm5_A |
205 |
344 |
142 |
1.70 |
9.86 |
37.942 |
7.905 |
T P |
| 3edt_B |
258 |
344 |
151 |
2.15 |
7.28 |
35.703 |
6.717 |
T P |
| 3ceq_B |
269 |
344 |
149 |
2.17 |
10.07 |
35.366 |
6.561 |
T P |
| 1ihg_A |
364 |
344 |
125 |
1.52 |
9.60 |
34.182 |
7.735 |
T P |
| 1p5q_A |
283 |
344 |
122 |
1.50 |
14.75 |
33.958 |
7.610 |
T P |
| 1qz2_A |
285 |
344 |
122 |
1.53 |
15.57 |
33.864 |
7.485 |
T P |
| 1kt1_A |
374 |
344 |
119 |
1.10 |
13.45 |
33.483 |
9.954 |
T P |
| 2c2l_A |
281 |
344 |
123 |
1.72 |
8.13 |
33.067 |
6.774 |
T P |
| 2j9q_A |
300 |
344 |
156 |
2.32 |
8.33 |
33.053 |
6.446 |
T P |
| 1na0_A |
119 |
344 |
118 |
1.18 |
16.10 |
32.834 |
9.196 |
T P |
| 2fbn_A |
153 |
344 |
118 |
1.47 |
8.47 |
32.381 |
7.527 |
T P |
| 1kt0_A |
357 |
344 |
115 |
1.10 |
13.04 |
32.377 |
9.566 |
T P |
| 1a17_A |
159 |
344 |
118 |
1.52 |
7.63 |
32.215 |
7.294 |
T P |
| 2dba_A |
148 |
344 |
121 |
1.65 |
8.26 |
32.211 |
6.932 |
T P |
| 2vyi_A |
128 |
344 |
117 |
1.55 |
9.40 |
31.991 |
7.091 |
T P |
| 1elr_A |
128 |
344 |
116 |
1.41 |
7.76 |
31.879 |
7.660 |
T P |
| 1elw_A |
117 |
344 |
116 |
1.32 |
7.76 |
31.720 |
8.142 |
T P |
| 1wao_1 |
471 |
344 |
130 |
2.14 |
6.15 |
31.567 |
5.797 |
T P |
| 2bug_A |
131 |
344 |
121 |
1.82 |
7.44 |
31.428 |
6.290 |
T P |
| 2if4_A |
258 |
344 |
113 |
2.16 |
7.96 |
26.451 |
5.009 |
T P |
| 1na3_A |
86 |
344 |
84 |
1.03 |
14.29 |
23.859 |
7.435 |
T P |
| 1ouv_A |
265 |
344 |
119 |
2.60 |
7.56 |
23.128 |
4.413 |
T P |
| 2avp_A |
68 |
344 |
68 |
0.88 |
16.18 |
19.496 |
6.911 |
T P |
| 1xi4_A |
1630 |
344 |
79 |
2.83 |
2.53 |
14.831 |
2.698 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]