LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_387.5wLII_11277_103
Total number of 3D structures: 111
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
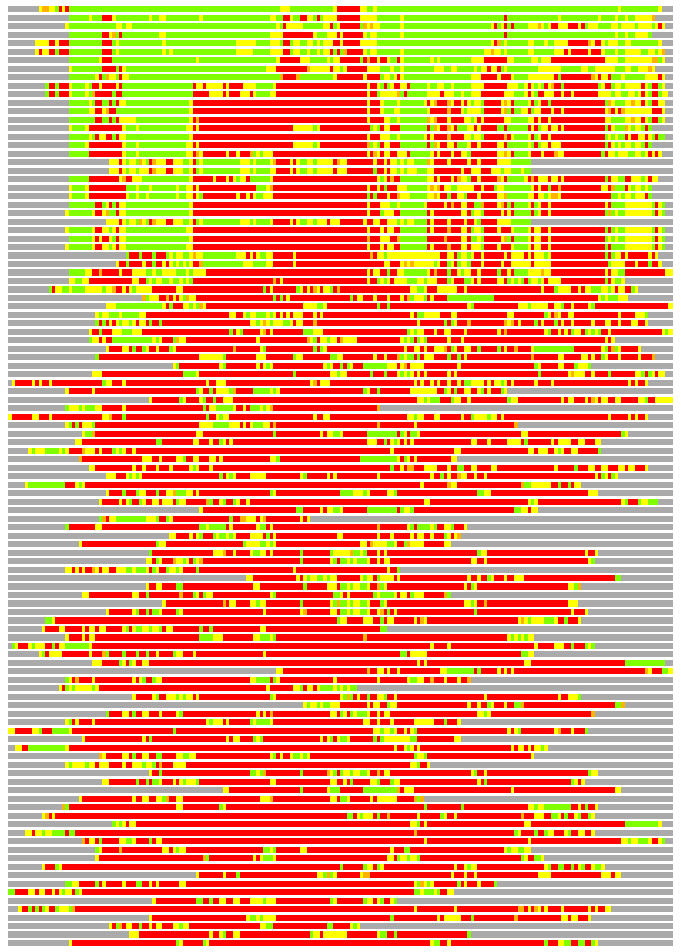
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1wek_A |
208 |
198 |
177 |
1.14 |
23.16 |
86.842 |
14.221 |
T P |
| 1ydh_A |
184 |
198 |
159 |
1.66 |
15.72 |
74.595 |
9.040 |
T P |
| 1weh_A |
171 |
198 |
161 |
1.86 |
18.63 |
73.144 |
8.203 |
T P |
| 2a33_A |
175 |
198 |
154 |
1.48 |
20.13 |
72.856 |
9.775 |
T P |
| 3gh1_A |
448 |
198 |
161 |
1.90 |
15.53 |
72.286 |
8.053 |
T P |
| 3bq9_A |
446 |
198 |
161 |
1.89 |
15.53 |
68.424 |
8.109 |
T P |
| 2iz6_A |
159 |
198 |
134 |
1.81 |
25.37 |
61.652 |
7.033 |
T P |
| 1t35_E |
184 |
198 |
152 |
1.95 |
17.76 |
59.882 |
7.406 |
T P |
| 1rcu_A |
170 |
198 |
138 |
1.86 |
24.64 |
59.794 |
7.042 |
T P |
| 3b51_X |
633 |
198 |
107 |
2.29 |
9.35 |
36.961 |
4.479 |
T P |
| 1su8_A |
633 |
198 |
109 |
2.34 |
6.42 |
36.744 |
4.476 |
T P |
| 1qpz_A |
339 |
198 |
84 |
2.18 |
8.33 |
31.440 |
3.684 |
T P |
| 1jhz_A |
276 |
198 |
82 |
2.31 |
7.32 |
31.090 |
3.399 |
T P |
| 1jh9_A |
339 |
198 |
83 |
2.12 |
8.43 |
31.047 |
3.741 |
T P |
| 1q7c_A |
243 |
198 |
87 |
2.21 |
12.64 |
30.752 |
3.774 |
T P |
| 2puc_A |
338 |
198 |
79 |
2.03 |
10.13 |
30.732 |
3.704 |
T P |
| 1q7b_A |
243 |
198 |
88 |
2.24 |
12.50 |
30.466 |
3.768 |
T P |
| 2cf2_E |
226 |
198 |
86 |
2.42 |
11.63 |
30.405 |
3.411 |
T P |
| 1jqb_A |
351 |
198 |
88 |
2.45 |
10.23 |
30.302 |
3.446 |
T P |
| 2b83_A |
351 |
198 |
91 |
2.49 |
9.89 |
30.237 |
3.516 |
T P |
| 2b4q_A |
256 |
198 |
87 |
2.36 |
14.94 |
30.217 |
3.539 |
T P |
| 2fwm_X |
212 |
198 |
86 |
2.35 |
15.12 |
30.160 |
3.514 |
T P |
| 1xg5_C |
257 |
198 |
86 |
2.38 |
10.47 |
29.809 |
3.471 |
T P |
| 1vpw_A |
338 |
198 |
82 |
2.12 |
10.98 |
29.809 |
3.699 |
T P |
| 1jft_A |
340 |
198 |
82 |
2.06 |
9.76 |
29.724 |
3.792 |
T P |
| 1kev_A |
351 |
198 |
88 |
2.46 |
10.23 |
29.720 |
3.431 |
T P |
| 2pue_A |
338 |
198 |
81 |
2.08 |
11.11 |
29.359 |
3.718 |
T P |
| 1jfs_A |
339 |
198 |
81 |
2.10 |
9.88 |
29.343 |
3.687 |
T P |
| 1bdh_A |
338 |
198 |
81 |
2.10 |
11.11 |
29.320 |
3.685 |
T P |
| 1h0c_A |
385 |
198 |
80 |
2.68 |
8.75 |
27.101 |
2.877 |
T P |
| 1j04_A |
386 |
198 |
76 |
2.75 |
9.21 |
26.744 |
2.666 |
T P |
| 2dr1_A |
381 |
198 |
72 |
2.54 |
9.72 |
24.174 |
2.723 |
T P |
| 2dkj_A |
402 |
198 |
69 |
2.46 |
13.04 |
23.297 |
2.692 |
T P |
| 1amu_A |
509 |
198 |
72 |
2.72 |
11.11 |
22.942 |
2.556 |
T P |
| 2q0t_A |
258 |
198 |
51 |
2.51 |
9.80 |
18.690 |
1.950 |
T P |
| 3dhi_A |
498 |
198 |
53 |
2.33 |
7.55 |
18.392 |
2.185 |
T P |
| 1t0q_A |
491 |
198 |
57 |
2.62 |
5.26 |
18.208 |
2.099 |
T P |
| 2rdb_A |
491 |
198 |
50 |
2.77 |
6.00 |
17.230 |
1.745 |
T P |
| 2inc_A |
491 |
198 |
50 |
2.63 |
6.00 |
16.906 |
1.833 |
T P |
| 3eqr_B |
271 |
198 |
48 |
2.48 |
8.33 |
16.716 |
1.860 |
T P |
| 2ew9_A |
149 |
198 |
50 |
2.59 |
12.00 |
16.543 |
1.859 |
T P |
| 1u46_B |
257 |
198 |
44 |
2.87 |
4.55 |
15.474 |
1.483 |
T P |
| 2gli_A |
155 |
198 |
40 |
2.43 |
7.50 |
14.181 |
1.580 |
T P |
| 1u54_A |
263 |
198 |
40 |
2.52 |
2.50 |
14.097 |
1.524 |
T P |
| 2inn_A |
496 |
198 |
40 |
2.88 |
2.50 |
13.711 |
1.341 |
T P |
| 1p6t_A |
151 |
198 |
40 |
2.59 |
7.50 |
13.382 |
1.489 |
T P |
| 1qup_A |
221 |
198 |
41 |
2.82 |
0.00 |
12.945 |
1.402 |
T P |
| 1kqk_A |
80 |
198 |
30 |
2.26 |
10.00 |
12.866 |
1.271 |
T P |
| 2inp_A |
494 |
198 |
37 |
3.02 |
0.00 |
12.712 |
1.187 |
T P |
| 1osd_A |
72 |
198 |
30 |
2.48 |
3.33 |
12.583 |
1.165 |
T P |
| 2cot_A |
77 |
198 |
31 |
2.16 |
6.45 |
12.516 |
1.374 |
T P |
| 2rml_A |
147 |
198 |
36 |
2.96 |
5.56 |
12.478 |
1.177 |
T P |
| 2prt_A |
115 |
198 |
36 |
2.62 |
2.78 |
12.123 |
1.323 |
T P |
| 1mwy_A |
73 |
198 |
33 |
2.36 |
12.12 |
12.043 |
1.343 |
T P |
| 1jk9_B |
243 |
198 |
36 |
2.94 |
5.56 |
11.851 |
1.186 |
T P |
| 2dlq_A |
124 |
198 |
35 |
2.77 |
2.86 |
11.806 |
1.221 |
T P |
| 2ee8_A |
106 |
198 |
32 |
2.44 |
9.38 |
11.568 |
1.261 |
T P |
| 1g2d_C |
89 |
198 |
36 |
2.56 |
0.00 |
11.490 |
1.354 |
T P |
| 1s6o_A |
76 |
198 |
34 |
2.46 |
8.82 |
11.384 |
1.329 |
T P |
| 2eq2_A |
46 |
198 |
28 |
2.24 |
3.57 |
11.265 |
1.195 |
T P |
| 1yju_A |
75 |
198 |
29 |
2.60 |
3.45 |
11.213 |
1.075 |
T P |
| 2gcf_A |
73 |
198 |
29 |
2.38 |
3.45 |
11.009 |
1.170 |
T P |
| 1y3j_A |
77 |
198 |
30 |
2.69 |
3.33 |
10.936 |
1.077 |
T P |
| 1yjr_A |
75 |
198 |
32 |
2.81 |
12.50 |
10.915 |
1.100 |
T P |
| 1a1g_A |
84 |
198 |
31 |
2.61 |
9.68 |
10.899 |
1.145 |
T P |
| 1aay_A |
85 |
198 |
32 |
2.71 |
6.25 |
10.857 |
1.138 |
T P |
| 1k0v_A |
73 |
198 |
29 |
2.58 |
6.90 |
10.857 |
1.084 |
T P |
| 2i13_A |
151 |
198 |
33 |
2.77 |
6.06 |
10.818 |
1.152 |
T P |
| 1a1h_A |
85 |
198 |
30 |
2.69 |
3.33 |
10.727 |
1.077 |
T P |
| 1opz_A |
76 |
198 |
32 |
2.67 |
6.25 |
10.659 |
1.154 |
T P |
| 1g2f_C |
89 |
198 |
32 |
2.70 |
0.00 |
10.653 |
1.144 |
T P |
| 1jk2_A |
85 |
198 |
30 |
2.54 |
3.33 |
10.628 |
1.138 |
T P |
| 2yt9_A |
95 |
198 |
29 |
2.67 |
6.90 |
10.581 |
1.047 |
T P |
| 1q8l_A |
84 |
198 |
27 |
2.55 |
0.00 |
10.564 |
1.020 |
T P |
| 2rop_A |
142 |
198 |
27 |
2.49 |
0.00 |
10.483 |
1.041 |
T P |
| 2dmd_A |
96 |
198 |
28 |
2.38 |
7.14 |
10.390 |
1.131 |
T P |
| 1kvi_A |
79 |
198 |
29 |
2.70 |
10.34 |
10.320 |
1.035 |
T P |
| 2ytk_A |
46 |
198 |
25 |
2.01 |
12.00 |
10.309 |
1.187 |
T P |
| 2eq1_A |
46 |
198 |
26 |
2.39 |
0.00 |
10.205 |
1.043 |
T P |
| 1aw0_A |
72 |
198 |
29 |
2.66 |
10.34 |
10.204 |
1.053 |
T P |
| 1jww_A |
80 |
198 |
29 |
2.45 |
3.45 |
10.170 |
1.138 |
T P |
| 1ubd_C |
114 |
198 |
30 |
2.68 |
6.67 |
10.150 |
1.081 |
T P |
| 1mey_F |
84 |
198 |
28 |
2.73 |
3.57 |
10.144 |
0.991 |
T P |
| 1a1i_A |
85 |
198 |
30 |
2.82 |
3.33 |
10.109 |
1.026 |
T P |
| 1fvq_A |
72 |
198 |
29 |
2.69 |
6.90 |
10.047 |
1.039 |
T P |
| 1f2i_G |
66 |
198 |
29 |
2.44 |
6.90 |
10.040 |
1.141 |
T P |
| 3cjk_B |
75 |
198 |
30 |
2.65 |
3.33 |
10.006 |
1.093 |
T P |
| 1x6e_A |
72 |
198 |
27 |
2.51 |
3.70 |
9.943 |
1.035 |
T P |
| 2qif_A |
69 |
198 |
29 |
2.62 |
3.45 |
9.938 |
1.067 |
T P |
| 1afi_A |
72 |
198 |
29 |
2.94 |
6.90 |
9.935 |
0.955 |
T P |
| 1zaa_C |
85 |
198 |
29 |
2.56 |
3.45 |
9.924 |
1.090 |
T P |
| 1p47_A |
87 |
198 |
31 |
2.66 |
3.23 |
9.855 |
1.121 |
T P |
| 2ene_A |
46 |
198 |
24 |
2.35 |
8.33 |
9.816 |
0.978 |
T P |
| 1cpz_A |
68 |
198 |
29 |
3.03 |
10.34 |
9.807 |
0.928 |
T P |
| 2emk_A |
46 |
198 |
24 |
2.16 |
4.17 |
9.800 |
1.063 |
T P |
| 1tf6_D |
182 |
198 |
28 |
2.82 |
0.00 |
9.777 |
0.960 |
T P |
| 2emw_A |
44 |
198 |
23 |
2.07 |
13.04 |
9.348 |
1.058 |
T P |
| 2ebt_A |
100 |
198 |
25 |
2.64 |
12.00 |
9.317 |
0.913 |
T P |
| 2ytr_A |
46 |
198 |
25 |
2.48 |
8.00 |
9.124 |
0.971 |
T P |
| 2g9o_A |
77 |
198 |
26 |
2.78 |
3.85 |
9.106 |
0.904 |
T P |
| 2ofg_X |
106 |
198 |
23 |
2.39 |
4.35 |
8.769 |
0.925 |
T P |
| 2en6_A |
46 |
198 |
25 |
2.39 |
16.00 |
8.725 |
1.005 |
T P |
| 2csh_A |
110 |
198 |
24 |
2.97 |
12.50 |
8.719 |
0.781 |
T P |
| 2aj0_A |
71 |
198 |
23 |
2.56 |
4.35 |
8.563 |
0.866 |
T P |
| 2yts_A |
46 |
198 |
23 |
2.25 |
4.35 |
8.264 |
0.978 |
T P |
| 2yrj_A |
46 |
198 |
25 |
2.71 |
12.00 |
8.258 |
0.891 |
T P |
| 2emj_A |
46 |
198 |
24 |
2.99 |
12.50 |
8.067 |
0.776 |
T P |
| 2eoe_A |
46 |
198 |
24 |
2.81 |
8.33 |
8.042 |
0.826 |
T P |
| 2em2_A |
46 |
198 |
23 |
2.64 |
13.04 |
7.863 |
0.840 |
T P |
| 2eog_A |
44 |
198 |
23 |
2.53 |
4.35 |
7.815 |
0.876 |
T P |
| 2eow_A |
46 |
198 |
18 |
2.01 |
22.22 |
7.315 |
0.852 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]