LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_391.5wLII_11277_114
Total number of 3D structures: 65
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
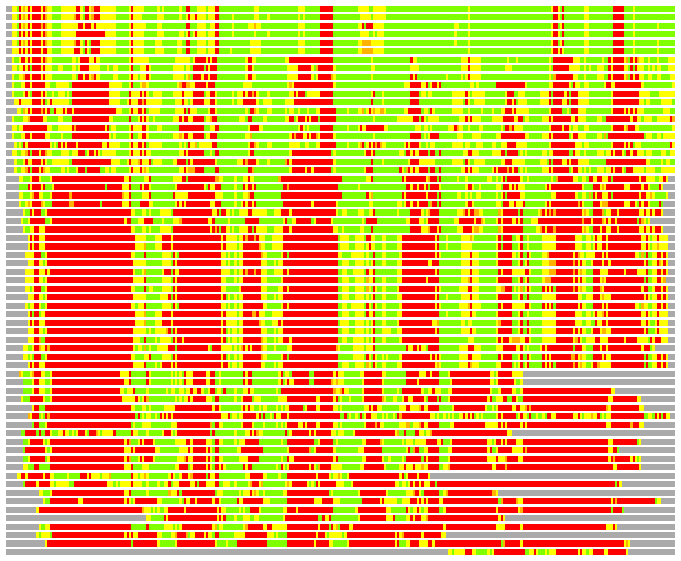
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qfg_A |
707 |
304 |
268 |
1.75 |
12.69 |
80.262 |
14.495 |
T P |
| 2fcp_A |
705 |
304 |
268 |
1.77 |
11.94 |
80.201 |
14.368 |
T P |
| 1by5_A |
697 |
304 |
265 |
1.80 |
12.83 |
79.105 |
13.923 |
T P |
| 1fi1_A |
707 |
304 |
261 |
1.69 |
12.26 |
78.666 |
14.582 |
T P |
| 1fcp_A |
705 |
304 |
268 |
1.86 |
12.31 |
78.403 |
13.679 |
T P |
| 1qff_A |
707 |
304 |
265 |
1.84 |
12.83 |
77.952 |
13.656 |
T P |
| 1kmp_A |
647 |
304 |
238 |
1.85 |
10.50 |
70.253 |
12.219 |
T P |
| 1po0_A |
661 |
304 |
248 |
1.96 |
10.89 |
68.938 |
12.011 |
T P |
| 1kmo_A |
661 |
304 |
244 |
1.90 |
11.48 |
66.600 |
12.179 |
T P |
| 2hdf_A |
584 |
304 |
203 |
1.95 |
7.39 |
57.722 |
9.905 |
T P |
| 1nqf_A |
544 |
304 |
222 |
2.16 |
6.31 |
56.141 |
9.834 |
T P |
| 2gsk_A |
590 |
304 |
210 |
2.09 |
6.19 |
55.410 |
9.597 |
T P |
| 2iah_A |
752 |
304 |
224 |
2.12 |
5.80 |
54.644 |
10.075 |
T P |
| 1fep_A |
680 |
304 |
210 |
2.13 |
7.14 |
54.520 |
9.400 |
T P |
| 1xkw_A |
655 |
304 |
220 |
2.03 |
9.09 |
54.203 |
10.342 |
T P |
| 2hdi_A |
598 |
304 |
204 |
1.97 |
8.33 |
53.966 |
9.853 |
T P |
| 1xkh_A |
687 |
304 |
223 |
2.15 |
6.28 |
52.405 |
9.918 |
T P |
| 3ddr_A |
753 |
304 |
218 |
2.03 |
8.72 |
52.354 |
10.237 |
T P |
| 2guf_A |
551 |
304 |
197 |
2.16 |
6.60 |
51.288 |
8.723 |
T P |
| 3csl_A |
753 |
304 |
219 |
2.10 |
9.13 |
50.907 |
9.949 |
T P |
| 1oh2_Q |
413 |
304 |
165 |
1.96 |
7.27 |
42.688 |
7.994 |
T P |
| 1af6_A |
421 |
304 |
157 |
1.87 |
11.46 |
42.026 |
7.954 |
T P |
| 1a0s_P |
413 |
304 |
167 |
2.02 |
7.19 |
41.922 |
7.883 |
T P |
| 2mpr_A |
421 |
304 |
164 |
2.05 |
7.93 |
38.883 |
7.642 |
T P |
| 2fgq_X |
330 |
304 |
145 |
2.42 |
9.66 |
35.540 |
5.764 |
T P |
| 2fgr_A |
332 |
304 |
136 |
2.10 |
5.88 |
35.498 |
6.176 |
T P |
| 1e54_A |
332 |
304 |
143 |
2.41 |
9.09 |
34.593 |
5.691 |
T P |
| 1gfm_A |
340 |
304 |
137 |
2.40 |
8.76 |
33.925 |
5.473 |
T P |
| 1gfq_A |
340 |
304 |
137 |
2.39 |
8.76 |
33.884 |
5.496 |
T P |
| 2j1n_A |
346 |
304 |
140 |
2.35 |
5.71 |
33.709 |
5.721 |
T P |
| 1bt9_A |
340 |
304 |
137 |
2.44 |
7.30 |
33.485 |
5.395 |
T P |
| 2ixw_A |
343 |
304 |
144 |
2.46 |
3.47 |
33.358 |
5.627 |
T P |
| 1hxt_A |
340 |
304 |
134 |
2.42 |
5.97 |
32.966 |
5.327 |
T P |
| 1hxu_A |
340 |
304 |
134 |
2.34 |
7.46 |
32.940 |
5.502 |
T P |
| 1mpf_A |
340 |
304 |
136 |
2.41 |
7.35 |
32.927 |
5.413 |
T P |
| 2ixx_A |
354 |
304 |
138 |
2.30 |
6.52 |
32.765 |
5.745 |
T P |
| 1hxx_A |
340 |
304 |
135 |
2.41 |
8.89 |
32.750 |
5.371 |
T P |
| 1gfo_A |
340 |
304 |
135 |
2.38 |
8.89 |
32.411 |
5.448 |
T P |
| 1gfn_A |
327 |
304 |
138 |
2.49 |
7.25 |
32.389 |
5.319 |
T P |
| 1osm_A |
342 |
304 |
144 |
2.47 |
6.94 |
32.187 |
5.613 |
T P |
| 2o4v_A |
411 |
304 |
140 |
2.35 |
7.86 |
32.057 |
5.718 |
T P |
| 1pho_A |
330 |
304 |
135 |
2.33 |
9.63 |
31.728 |
5.565 |
T P |
| 2zfg_A |
339 |
304 |
130 |
2.41 |
7.69 |
30.892 |
5.179 |
T P |
| 1t16_A |
427 |
304 |
119 |
2.18 |
4.20 |
28.625 |
5.229 |
T P |
| 2r4p_A |
418 |
304 |
115 |
2.06 |
4.35 |
28.107 |
5.314 |
T P |
| 3dwo_X |
444 |
304 |
123 |
2.20 |
4.88 |
28.081 |
5.340 |
T P |
| 2iwv_A |
277 |
304 |
117 |
2.18 |
3.42 |
27.766 |
5.137 |
T P |
| 3dwn_A |
421 |
304 |
119 |
2.26 |
7.56 |
27.524 |
5.037 |
T P |
| 1gfp_A |
340 |
304 |
127 |
2.45 |
7.09 |
27.446 |
4.983 |
T P |
| 2r4l_B |
421 |
304 |
119 |
2.33 |
7.56 |
27.208 |
4.906 |
T P |
| 2r4o_A |
421 |
304 |
113 |
2.16 |
7.96 |
26.841 |
5.001 |
T P |
| 2r4n_A |
421 |
304 |
116 |
2.28 |
2.59 |
26.821 |
4.880 |
T P |
| 2r89_A |
363 |
304 |
103 |
2.12 |
11.65 |
26.596 |
4.634 |
T P |
| 2r88_A |
365 |
304 |
108 |
2.27 |
3.70 |
26.337 |
4.555 |
T P |
| 3bs0_A |
414 |
304 |
114 |
2.32 |
7.02 |
26.061 |
4.704 |
T P |
| 2r8a_A |
360 |
304 |
103 |
2.25 |
9.71 |
24.199 |
4.378 |
T P |
| 3bry_A |
389 |
304 |
109 |
2.43 |
8.26 |
24.063 |
4.306 |
T P |
| 1i78_A |
297 |
304 |
98 |
2.08 |
5.10 |
24.007 |
4.490 |
T P |
| 2jqy_A |
280 |
304 |
91 |
2.49 |
5.49 |
21.115 |
3.515 |
T P |
| 1bxw_A |
172 |
304 |
78 |
2.20 |
3.85 |
19.876 |
3.394 |
T P |
| 1qj8_A |
148 |
304 |
71 |
2.05 |
8.45 |
18.243 |
3.303 |
T P |
| 1g90_A |
176 |
304 |
69 |
2.46 |
5.80 |
16.550 |
2.694 |
T P |
| 2ge4_A |
177 |
304 |
66 |
2.54 |
12.12 |
15.031 |
2.501 |
T P |
| 2jmm_A |
156 |
304 |
57 |
2.11 |
12.28 |
13.896 |
2.580 |
T P |
| 2z3q_B |
81 |
304 |
40 |
2.53 |
2.50 |
8.581 |
1.520 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]