LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_393.5wLII_11277_121
Total number of 3D structures: 94
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
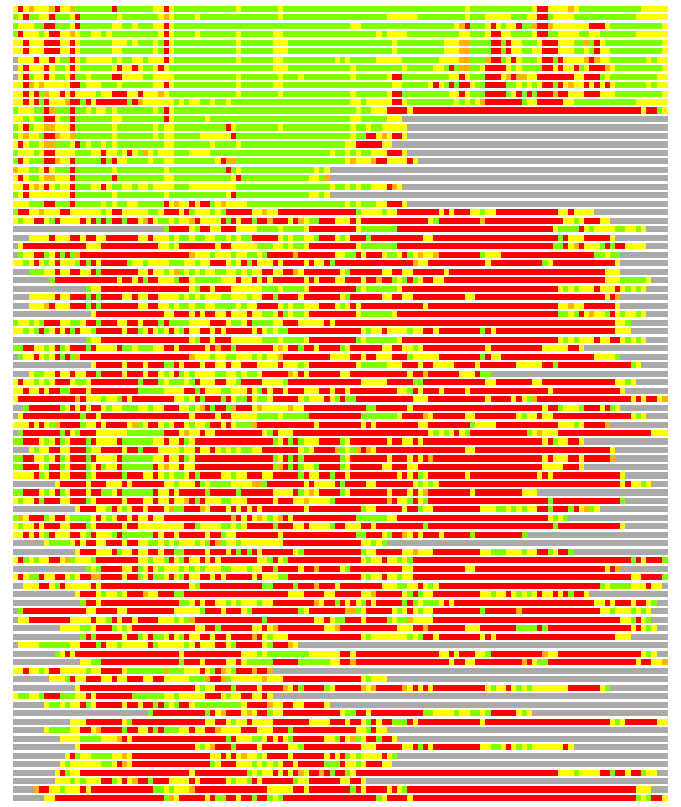
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2awn_B |
374 |
126 |
120 |
1.75 |
17.50 |
88.234 |
6.488 |
T P |
| 2r6g_B |
372 |
126 |
121 |
1.83 |
18.18 |
85.815 |
6.275 |
T P |
| 2it1_A |
362 |
126 |
114 |
1.77 |
22.81 |
83.546 |
6.086 |
T P |
| 1q1b_A |
367 |
126 |
118 |
1.98 |
18.64 |
81.061 |
5.671 |
T P |
| 1v43_A |
353 |
126 |
113 |
1.93 |
22.12 |
80.228 |
5.562 |
T P |
| 1vci_A |
353 |
126 |
113 |
1.91 |
22.12 |
80.132 |
5.613 |
T P |
| 1g29_1 |
372 |
126 |
114 |
2.15 |
24.56 |
74.325 |
5.057 |
T P |
| 2yyz_A |
358 |
126 |
107 |
1.94 |
24.30 |
72.883 |
5.238 |
T P |
| 1oxx_K |
352 |
126 |
103 |
1.92 |
17.48 |
71.439 |
5.088 |
T P |
| 2d62_A |
375 |
126 |
108 |
2.00 |
22.22 |
69.426 |
5.136 |
T P |
| 1oxs_C |
352 |
126 |
101 |
1.94 |
14.85 |
69.218 |
4.946 |
T P |
| 1z47_A |
345 |
126 |
99 |
2.15 |
18.18 |
62.550 |
4.398 |
T P |
| 3d31_A |
348 |
126 |
73 |
1.72 |
26.03 |
53.543 |
4.004 |
T P |
| 2onk_A |
240 |
126 |
71 |
1.57 |
22.54 |
53.401 |
4.241 |
T P |
| 2hyd_A |
578 |
126 |
73 |
1.98 |
19.18 |
50.966 |
3.509 |
T P |
| 2cbz_A |
230 |
126 |
69 |
1.95 |
17.39 |
48.874 |
3.373 |
T P |
| 1vpl_A |
238 |
126 |
66 |
1.71 |
19.70 |
48.264 |
3.638 |
T P |
| 1b0u_A |
258 |
126 |
69 |
2.09 |
24.64 |
45.296 |
3.154 |
T P |
| 2ihy_B |
261 |
126 |
68 |
2.10 |
22.06 |
45.098 |
3.086 |
T P |
| 1f3o_A |
232 |
126 |
58 |
1.55 |
18.97 |
43.059 |
3.512 |
T P |
| 2pcj_A |
223 |
126 |
57 |
1.85 |
15.79 |
42.711 |
2.928 |
T P |
| 2olj_A |
242 |
126 |
70 |
2.31 |
24.29 |
41.753 |
2.902 |
T P |
| 1l2t_B |
232 |
126 |
58 |
1.87 |
17.24 |
41.229 |
2.938 |
T P |
| 3c41_J |
242 |
126 |
66 |
2.03 |
19.70 |
39.510 |
3.095 |
T P |
| 2vr5_A |
715 |
126 |
57 |
2.90 |
5.26 |
27.604 |
1.897 |
T P |
| 1qho_A |
686 |
126 |
50 |
2.68 |
8.00 |
25.912 |
1.800 |
T P |
| 1p9v_A |
310 |
126 |
44 |
2.19 |
6.82 |
25.810 |
1.923 |
T P |
| 1v3j_A |
686 |
126 |
50 |
2.62 |
0.00 |
25.205 |
1.841 |
T P |
| 1s7j_A |
260 |
126 |
45 |
2.36 |
11.11 |
24.575 |
1.832 |
T P |
| 1v3k_A |
686 |
126 |
48 |
2.63 |
8.33 |
24.402 |
1.756 |
T P |
| 1ukt_A |
686 |
126 |
48 |
2.68 |
4.17 |
24.339 |
1.730 |
T P |
| 1cgu_A |
684 |
126 |
47 |
2.78 |
10.64 |
24.321 |
1.631 |
T P |
| 3edn_A |
299 |
126 |
43 |
2.61 |
0.00 |
24.185 |
1.589 |
T P |
| 1u1v_A |
278 |
126 |
45 |
2.57 |
2.22 |
24.117 |
1.685 |
T P |
| 3cgt_A |
684 |
126 |
45 |
2.68 |
13.33 |
24.100 |
1.618 |
T P |
| 1cyg_A |
680 |
126 |
49 |
2.72 |
2.04 |
24.045 |
1.735 |
T P |
| 1qy9_D |
297 |
126 |
43 |
2.49 |
6.98 |
24.000 |
1.663 |
T P |
| 1uks_A |
686 |
126 |
43 |
2.54 |
6.98 |
23.904 |
1.628 |
T P |
| 2d0f_A |
637 |
126 |
47 |
2.61 |
4.26 |
23.840 |
1.736 |
T P |
| 1xub_A |
278 |
126 |
44 |
2.55 |
2.27 |
23.827 |
1.662 |
T P |
| 1ji2_A |
585 |
126 |
45 |
2.81 |
13.33 |
23.688 |
1.549 |
T P |
| 1d7f_A |
686 |
126 |
49 |
2.78 |
8.16 |
23.616 |
1.700 |
T P |
| 1t6k_A |
278 |
126 |
43 |
2.74 |
6.98 |
23.413 |
1.514 |
T P |
| 1cgt_A |
684 |
126 |
43 |
2.72 |
9.30 |
23.410 |
1.524 |
T P |
| 2e8y_A |
712 |
126 |
45 |
2.74 |
8.89 |
23.395 |
1.583 |
T P |
| 2pwf_C |
556 |
126 |
43 |
2.72 |
0.00 |
23.178 |
1.525 |
T P |
| 2otn_A |
293 |
126 |
44 |
2.64 |
6.82 |
23.176 |
1.608 |
T P |
| 1ym5_A |
294 |
126 |
47 |
2.71 |
4.26 |
23.133 |
1.670 |
T P |
| 1u1w_B |
279 |
126 |
41 |
2.36 |
2.44 |
23.091 |
1.663 |
T P |
| 1m7x_B |
591 |
126 |
45 |
2.93 |
6.67 |
22.867 |
1.486 |
T P |
| 2q9j_A |
274 |
126 |
44 |
2.57 |
2.27 |
22.785 |
1.646 |
T P |
| 1wzk_A |
585 |
126 |
42 |
2.68 |
9.52 |
22.628 |
1.513 |
T P |
| 1pam_A |
686 |
126 |
42 |
2.79 |
4.76 |
22.574 |
1.452 |
T P |
| 1jf6_A |
585 |
126 |
41 |
2.73 |
12.20 |
22.484 |
1.447 |
T P |
| 1wzl_A |
585 |
126 |
41 |
2.64 |
12.20 |
22.407 |
1.495 |
T P |
| 1zja_A |
557 |
126 |
40 |
2.74 |
5.00 |
21.969 |
1.406 |
T P |
| 1u0k_A |
284 |
126 |
43 |
2.83 |
6.98 |
21.965 |
1.467 |
T P |
| 1g1y_A |
585 |
126 |
40 |
2.60 |
12.50 |
21.965 |
1.482 |
T P |
| 2pwe_A |
556 |
126 |
43 |
2.85 |
6.98 |
21.875 |
1.456 |
T P |
| 2d2o_A |
585 |
126 |
43 |
2.75 |
6.98 |
21.675 |
1.510 |
T P |
| 1bf2_A |
750 |
126 |
40 |
2.58 |
12.50 |
21.513 |
1.494 |
T P |
| 2pwh_A |
556 |
126 |
44 |
2.94 |
4.55 |
21.488 |
1.449 |
T P |
| 1vfo_A |
585 |
126 |
37 |
2.65 |
5.41 |
21.430 |
1.343 |
T P |
| 2hy5_A |
130 |
126 |
42 |
2.62 |
9.52 |
21.268 |
1.544 |
T P |
| 1sma_A |
588 |
126 |
42 |
2.61 |
9.52 |
21.190 |
1.551 |
T P |
| 1izk_A |
637 |
126 |
42 |
2.60 |
0.00 |
21.130 |
1.556 |
T P |
| 4cgt_A |
678 |
126 |
39 |
2.72 |
10.26 |
20.908 |
1.381 |
T P |
| 1uh4_A |
637 |
126 |
41 |
2.74 |
4.88 |
20.553 |
1.442 |
T P |
| 1jf5_A |
585 |
126 |
39 |
2.77 |
10.26 |
20.360 |
1.359 |
T P |
| 1j0j_A |
588 |
126 |
40 |
2.76 |
12.50 |
20.188 |
1.399 |
T P |
| 3fve_A |
283 |
126 |
38 |
2.70 |
7.89 |
20.104 |
1.358 |
T P |
| 1qya_B |
307 |
126 |
39 |
2.72 |
5.13 |
20.085 |
1.381 |
T P |
| 2azp_A |
318 |
126 |
39 |
2.88 |
15.38 |
19.705 |
1.310 |
T P |
| 1bwz_A |
274 |
126 |
37 |
2.74 |
10.81 |
19.609 |
1.305 |
T P |
| 1ji1_A |
637 |
126 |
38 |
2.75 |
2.63 |
19.548 |
1.333 |
T P |
| 2hy5_C |
101 |
126 |
38 |
2.66 |
0.00 |
19.509 |
1.378 |
T P |
| 3ejx_D |
301 |
126 |
36 |
2.44 |
16.67 |
19.497 |
1.418 |
T P |
| 2gke_A |
274 |
126 |
34 |
2.52 |
5.88 |
19.478 |
1.300 |
T P |
| 2d1p_C |
95 |
126 |
36 |
2.68 |
2.78 |
19.463 |
1.296 |
T P |
| 1x9a_A |
87 |
126 |
38 |
2.91 |
13.16 |
19.193 |
1.264 |
T P |
| 1gvi_A |
588 |
126 |
36 |
2.83 |
11.11 |
19.052 |
1.229 |
T P |
| 2d1p_A |
130 |
126 |
33 |
2.47 |
9.09 |
18.205 |
1.285 |
T P |
| 1jx7_A |
117 |
126 |
32 |
2.48 |
9.38 |
17.841 |
1.239 |
T P |
| 1wzm_A |
585 |
126 |
30 |
2.49 |
10.00 |
17.788 |
1.156 |
T P |
| 1w61_B |
357 |
126 |
33 |
2.96 |
12.12 |
17.705 |
1.079 |
T P |
| 1l1s_A |
111 |
126 |
34 |
2.64 |
11.76 |
17.564 |
1.243 |
T P |
| 2qs7_A |
138 |
126 |
32 |
2.63 |
9.38 |
17.527 |
1.173 |
T P |
| 1j0h_A |
588 |
126 |
33 |
2.60 |
9.09 |
17.442 |
1.224 |
T P |
| 2hy5_B |
132 |
126 |
33 |
2.78 |
6.06 |
17.017 |
1.144 |
T P |
| 2d1p_B |
119 |
126 |
34 |
2.68 |
8.82 |
16.977 |
1.221 |
T P |
| 2q9h_A |
274 |
126 |
31 |
2.72 |
0.00 |
16.761 |
1.098 |
T P |
| 1rhx_A |
87 |
126 |
31 |
2.82 |
3.23 |
16.664 |
1.063 |
T P |
| 1izj_A |
637 |
126 |
30 |
2.79 |
0.00 |
16.022 |
1.037 |
T P |
| 1ea9_C |
583 |
126 |
20 |
2.42 |
10.00 |
11.713 |
0.792 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]