LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_395.5wLII_11277_123
Total number of 3D structures: 114
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
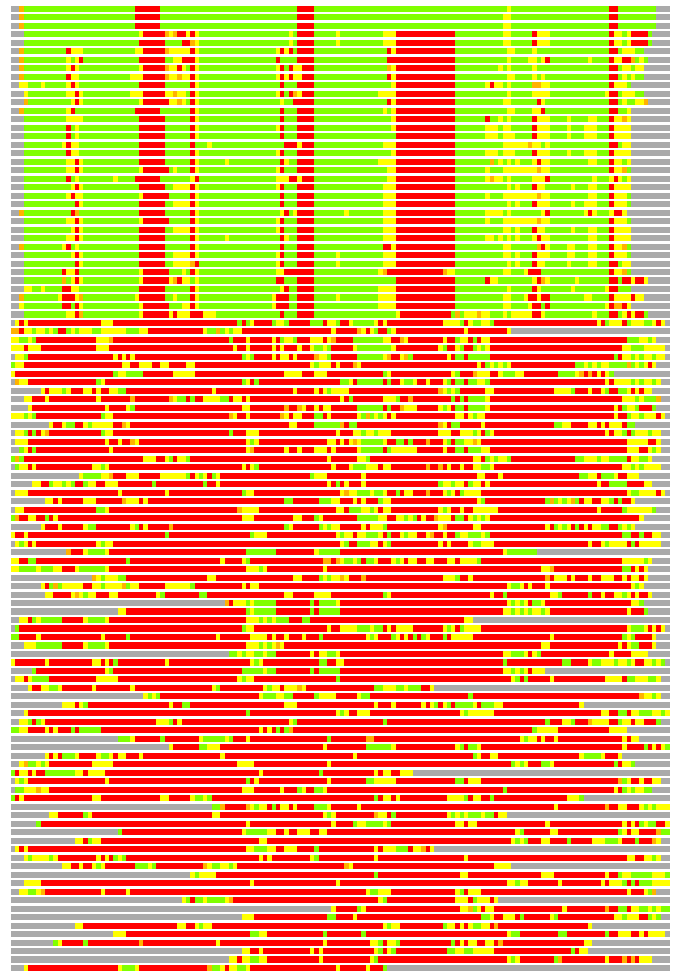
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3c99_A |
380 |
154 |
137 |
0.68 |
20.44 |
88.249 |
17.582 |
T P |
| 3cfs_B |
383 |
154 |
137 |
0.82 |
20.44 |
88.044 |
14.848 |
T P |
| 3cfv_B |
393 |
154 |
137 |
0.84 |
20.44 |
87.790 |
14.545 |
T P |
| 2aq5_A |
395 |
154 |
115 |
1.45 |
10.43 |
72.015 |
7.437 |
T P |
| 2b4e_A |
386 |
154 |
115 |
1.34 |
10.43 |
71.992 |
7.968 |
T P |
| 2pm6_D |
288 |
154 |
116 |
1.58 |
10.34 |
70.979 |
6.922 |
T P |
| 3fm0_A |
328 |
154 |
112 |
1.50 |
16.07 |
70.682 |
6.999 |
T P |
| 2pm9_B |
280 |
154 |
117 |
1.69 |
9.40 |
70.566 |
6.554 |
T P |
| 2pm7_D |
288 |
154 |
117 |
1.62 |
9.40 |
70.261 |
6.792 |
T P |
| 1erj_C |
357 |
154 |
114 |
1.47 |
11.40 |
69.993 |
7.238 |
T P |
| 3bg1_A |
285 |
154 |
117 |
1.75 |
9.40 |
69.651 |
6.323 |
T P |
| 2hes_X |
308 |
154 |
115 |
1.63 |
10.43 |
69.575 |
6.658 |
T P |
| 1vyh_C |
310 |
154 |
113 |
1.38 |
13.27 |
69.330 |
7.653 |
T P |
| 2zkq_a |
306 |
154 |
113 |
1.52 |
9.73 |
69.008 |
6.969 |
T P |
| 1tbg_A |
340 |
154 |
113 |
1.47 |
11.50 |
68.840 |
7.218 |
T P |
| 2bcj_B |
339 |
154 |
113 |
1.45 |
11.50 |
68.774 |
7.271 |
T P |
| 2pbi_D |
354 |
154 |
112 |
1.55 |
13.39 |
68.714 |
6.776 |
T P |
| 1got_B |
339 |
154 |
113 |
1.48 |
11.50 |
68.657 |
7.158 |
T P |
| 2h9l_A |
321 |
154 |
113 |
1.50 |
11.50 |
68.404 |
7.041 |
T P |
| 2h6n_B |
305 |
154 |
114 |
1.70 |
7.89 |
68.193 |
6.317 |
T P |
| 1gxr_A |
335 |
154 |
113 |
1.53 |
9.73 |
68.179 |
6.917 |
T P |
| 2cnx_A |
306 |
154 |
113 |
1.55 |
10.62 |
68.170 |
6.834 |
T P |
| 3emh_A |
300 |
154 |
114 |
1.69 |
7.02 |
68.153 |
6.366 |
T P |
| 2h14_A |
303 |
154 |
113 |
1.56 |
10.62 |
68.099 |
6.804 |
T P |
| 3frx_B |
313 |
154 |
111 |
1.47 |
12.61 |
68.091 |
7.061 |
T P |
| 2h9m_A |
304 |
154 |
114 |
1.72 |
7.89 |
68.045 |
6.277 |
T P |
| 2co0_A |
304 |
154 |
113 |
1.57 |
10.62 |
68.037 |
6.768 |
T P |
| 2gnq_A |
316 |
154 |
113 |
1.54 |
11.50 |
67.965 |
6.891 |
T P |
| 3dm0_A |
675 |
154 |
112 |
1.54 |
14.29 |
67.934 |
6.825 |
T P |
| 2g99_A |
304 |
154 |
113 |
1.61 |
10.62 |
67.860 |
6.589 |
T P |
| 2g9a_A |
310 |
154 |
112 |
1.55 |
11.61 |
67.849 |
6.774 |
T P |
| 1nex_B |
444 |
154 |
109 |
1.62 |
13.76 |
67.384 |
6.354 |
T P |
| 1r5m_A |
351 |
154 |
110 |
1.60 |
8.18 |
67.159 |
6.475 |
T P |
| 1a0r_B |
339 |
154 |
111 |
1.47 |
12.61 |
66.943 |
7.079 |
T P |
| 1p22_A |
402 |
154 |
109 |
1.68 |
13.76 |
65.166 |
6.108 |
T P |
| 2ovr_B |
442 |
154 |
107 |
1.59 |
11.21 |
64.750 |
6.336 |
T P |
| 1nr0_A |
610 |
154 |
111 |
1.85 |
9.91 |
62.702 |
5.691 |
T P |
| 2huf_A |
384 |
154 |
50 |
2.55 |
14.00 |
21.669 |
1.886 |
T P |
| 2fgs_A |
169 |
154 |
47 |
2.67 |
6.38 |
21.574 |
1.695 |
T P |
| 1p3w_B |
385 |
154 |
47 |
2.54 |
10.64 |
21.360 |
1.784 |
T P |
| 1eg5_B |
365 |
154 |
47 |
2.57 |
2.13 |
20.488 |
1.758 |
T P |
| 1j04_A |
386 |
154 |
47 |
2.52 |
6.38 |
20.372 |
1.792 |
T P |
| 2gms_A |
390 |
154 |
45 |
2.65 |
2.22 |
19.170 |
1.638 |
T P |
| 2rdo_7 |
684 |
154 |
44 |
2.56 |
2.27 |
19.111 |
1.652 |
T P |
| 1vjo_A |
377 |
154 |
44 |
2.46 |
4.55 |
19.051 |
1.717 |
T P |
| 2dr1_A |
381 |
154 |
43 |
2.88 |
9.30 |
18.988 |
1.445 |
T P |
| 1dfo_B |
417 |
154 |
42 |
2.60 |
4.76 |
18.744 |
1.553 |
T P |
| 1m32_B |
362 |
154 |
39 |
2.56 |
5.13 |
18.294 |
1.468 |
T P |
| 2e7j_A |
344 |
154 |
42 |
2.76 |
7.14 |
18.075 |
1.468 |
T P |
| 1mdx_A |
366 |
154 |
41 |
2.58 |
9.76 |
18.048 |
1.529 |
T P |
| 1t3i_A |
406 |
154 |
44 |
2.80 |
11.36 |
18.005 |
1.516 |
T P |
| 1b5p_A |
382 |
154 |
40 |
2.68 |
0.00 |
17.924 |
1.438 |
T P |
| 3f9t_A |
394 |
154 |
39 |
2.74 |
2.56 |
17.900 |
1.373 |
T P |
| 2r0t_A |
386 |
154 |
40 |
2.37 |
2.50 |
17.793 |
1.617 |
T P |
| 1i29_A |
405 |
154 |
39 |
2.69 |
7.69 |
17.723 |
1.399 |
T P |
| 1g0d_A |
666 |
154 |
42 |
2.60 |
11.90 |
17.539 |
1.557 |
T P |
| 1v2d_A |
365 |
154 |
38 |
2.54 |
0.00 |
17.317 |
1.439 |
T P |
| 1bkg_A |
382 |
154 |
39 |
2.76 |
2.56 |
17.245 |
1.364 |
T P |
| 3b8x_B |
388 |
154 |
40 |
2.70 |
2.50 |
17.142 |
1.429 |
T P |
| 1kmj_A |
405 |
154 |
40 |
2.70 |
10.00 |
17.064 |
1.429 |
T P |
| 1n2t_A |
385 |
154 |
38 |
2.65 |
5.26 |
16.911 |
1.379 |
T P |
| 3frk_A |
365 |
154 |
40 |
2.71 |
2.50 |
16.736 |
1.425 |
T P |
| 2z9v_A |
392 |
154 |
38 |
2.42 |
2.63 |
16.671 |
1.510 |
T P |
| 1jf9_A |
405 |
154 |
38 |
2.76 |
7.89 |
16.667 |
1.328 |
T P |
| 2ene_A |
46 |
154 |
28 |
1.81 |
3.57 |
16.609 |
1.463 |
T P |
| 2ch1_A |
387 |
154 |
40 |
2.72 |
2.50 |
16.595 |
1.416 |
T P |
| 1ubd_C |
114 |
154 |
33 |
2.40 |
15.15 |
16.428 |
1.319 |
T P |
| 2yrr_A |
352 |
154 |
38 |
2.74 |
5.26 |
16.133 |
1.337 |
T P |
| 1h0c_A |
385 |
154 |
39 |
2.90 |
2.56 |
16.115 |
1.302 |
T P |
| 3b46_B |
424 |
154 |
35 |
2.78 |
8.57 |
16.069 |
1.215 |
T P |
| 2eok_A |
42 |
154 |
28 |
2.13 |
7.14 |
15.673 |
1.257 |
T P |
| 2eog_A |
44 |
154 |
28 |
2.08 |
3.57 |
15.573 |
1.283 |
T P |
| 2yrj_A |
46 |
154 |
29 |
2.21 |
10.34 |
15.447 |
1.254 |
T P |
| 1b5o_A |
382 |
154 |
35 |
2.57 |
5.71 |
15.260 |
1.312 |
T P |
| 1ecx_B |
365 |
154 |
34 |
2.53 |
14.71 |
15.168 |
1.295 |
T P |
| 2epy_A |
42 |
154 |
31 |
2.23 |
12.90 |
15.044 |
1.333 |
T P |
| 2ytr_A |
46 |
154 |
30 |
2.46 |
3.33 |
14.539 |
1.174 |
T P |
| 1qz9_A |
404 |
154 |
33 |
2.49 |
0.00 |
14.481 |
1.274 |
T P |
| 2en6_A |
46 |
154 |
26 |
2.04 |
3.85 |
14.384 |
1.217 |
T P |
| 2csh_A |
110 |
154 |
33 |
2.62 |
0.00 |
13.799 |
1.212 |
T P |
| 2yts_A |
46 |
154 |
31 |
2.73 |
9.68 |
13.278 |
1.095 |
T P |
| 2emh_A |
46 |
154 |
26 |
2.34 |
3.85 |
12.965 |
1.066 |
T P |
| 1a1h_A |
85 |
154 |
29 |
2.66 |
0.00 |
12.528 |
1.050 |
T P |
| 2ytn_A |
46 |
154 |
29 |
2.55 |
6.90 |
12.515 |
1.096 |
T P |
| 2hdy_A |
403 |
154 |
28 |
2.79 |
0.00 |
12.511 |
0.967 |
T P |
| 1mey_F |
84 |
154 |
27 |
2.61 |
7.41 |
12.467 |
0.998 |
T P |
| 1g2d_C |
89 |
154 |
26 |
2.51 |
0.00 |
12.429 |
0.996 |
T P |
| 2eoe_A |
46 |
154 |
24 |
2.18 |
8.33 |
12.403 |
1.055 |
T P |
| 1c0n_A |
404 |
154 |
26 |
2.66 |
11.54 |
12.400 |
0.943 |
T P |
| 2em2_A |
46 |
154 |
28 |
2.45 |
7.14 |
12.369 |
1.098 |
T P |
| 1a1i_A |
85 |
154 |
25 |
2.40 |
4.00 |
12.323 |
1.000 |
T P |
| 2eq1_A |
46 |
154 |
25 |
2.74 |
4.00 |
12.318 |
0.880 |
T P |
| 1elu_A |
382 |
154 |
26 |
2.53 |
19.23 |
12.317 |
0.988 |
T P |
| 3cai_A |
396 |
154 |
27 |
2.80 |
7.41 |
11.933 |
0.930 |
T P |
| 2en0_A |
42 |
154 |
28 |
2.75 |
10.71 |
11.875 |
0.981 |
T P |
| 2enf_A |
46 |
154 |
26 |
2.62 |
0.00 |
11.864 |
0.956 |
T P |
| 2en4_A |
46 |
154 |
24 |
2.43 |
8.33 |
11.845 |
0.950 |
T P |
| 2eol_A |
42 |
154 |
26 |
2.48 |
11.54 |
11.611 |
1.009 |
T P |
| 2yu8_A |
46 |
154 |
24 |
2.55 |
8.33 |
11.066 |
0.904 |
T P |
| 2eme_A |
46 |
154 |
24 |
2.59 |
4.17 |
11.063 |
0.892 |
T P |
| 1x6e_A |
72 |
154 |
25 |
2.70 |
4.00 |
10.916 |
0.894 |
T P |
| 2epz_A |
46 |
154 |
25 |
2.73 |
4.00 |
10.906 |
0.883 |
T P |
| 1v65_A |
64 |
154 |
25 |
2.54 |
4.00 |
10.845 |
0.946 |
T P |
| 2yti_A |
46 |
154 |
25 |
2.74 |
0.00 |
10.580 |
0.881 |
T P |
| 2yrh_A |
44 |
154 |
23 |
2.78 |
0.00 |
10.444 |
0.799 |
T P |
| 2eow_A |
46 |
154 |
21 |
2.43 |
9.52 |
10.422 |
0.830 |
T P |
| 2emw_A |
44 |
154 |
23 |
2.76 |
0.00 |
10.253 |
0.804 |
T P |
| 2eqw_A |
42 |
154 |
24 |
2.54 |
12.50 |
10.134 |
0.910 |
T P |
| 2eq3_A |
46 |
154 |
23 |
2.83 |
4.35 |
10.096 |
0.785 |
T P |
| 2emv_A |
44 |
154 |
23 |
2.57 |
8.70 |
10.032 |
0.863 |
T P |
| 2ep3_A |
46 |
154 |
21 |
2.76 |
0.00 |
9.697 |
0.734 |
T P |
| 2cot_A |
77 |
154 |
23 |
2.77 |
0.00 |
9.328 |
0.801 |
T P |
| 2em1_A |
44 |
154 |
22 |
2.94 |
4.55 |
9.095 |
0.723 |
T P |
| 2emp_A |
46 |
154 |
18 |
2.69 |
5.56 |
8.949 |
0.646 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]