LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_40.5wLII_10850_11
Total number of 3D structures: 34
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
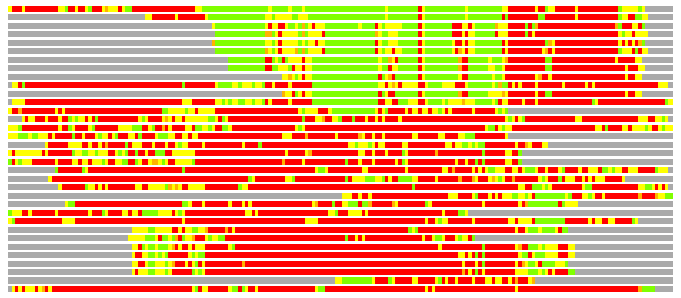
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3dwk_C |
623 |
199 |
122 |
1.81 |
10.66 |
55.417 |
6.400 |
T P |
| 2olv_B |
618 |
199 |
102 |
1.62 |
10.78 |
47.730 |
5.939 |
T P |
| 2v2f_F |
366 |
199 |
87 |
2.10 |
8.05 |
36.936 |
3.956 |
T P |
| 2c5w_B |
385 |
199 |
90 |
2.15 |
11.11 |
35.995 |
3.993 |
T P |
| 2zc6_B |
384 |
199 |
89 |
2.10 |
10.11 |
35.632 |
4.040 |
T P |
| 2c6w_B |
384 |
199 |
90 |
2.19 |
11.11 |
35.537 |
3.928 |
T P |
| 2bg1_A |
455 |
199 |
81 |
1.83 |
7.41 |
34.948 |
4.194 |
T P |
| 2fff_B |
453 |
199 |
80 |
1.88 |
7.50 |
33.102 |
4.042 |
T P |
| 2uwx_A |
468 |
199 |
67 |
1.92 |
7.46 |
29.797 |
3.324 |
T P |
| 3equ_A |
429 |
199 |
82 |
2.30 |
8.54 |
29.412 |
3.417 |
T P |
| 2uwy_A |
469 |
199 |
66 |
1.97 |
9.09 |
29.140 |
3.195 |
T P |
| 3eqv_A |
443 |
199 |
82 |
2.32 |
12.20 |
28.566 |
3.383 |
T P |
| 1h6k_C |
733 |
199 |
57 |
2.65 |
3.51 |
19.158 |
2.073 |
T P |
| 1bg1_A |
559 |
199 |
58 |
2.86 |
8.62 |
18.607 |
1.958 |
T P |
| 2wb7_A |
525 |
199 |
56 |
2.60 |
7.14 |
17.931 |
2.073 |
T P |
| 2hko_A |
647 |
199 |
53 |
2.77 |
7.55 |
17.652 |
1.845 |
T P |
| 2h94_A |
647 |
199 |
49 |
2.83 |
4.08 |
17.379 |
1.675 |
T P |
| 2z3y_A |
643 |
199 |
51 |
2.77 |
1.96 |
16.954 |
1.776 |
T P |
| 2dw4_A |
634 |
199 |
49 |
2.65 |
0.00 |
16.776 |
1.781 |
T P |
| 1qvr_A |
803 |
199 |
49 |
2.64 |
8.16 |
16.124 |
1.789 |
T P |
| 2b5u_A |
470 |
199 |
47 |
2.72 |
8.51 |
15.623 |
1.669 |
T P |
| 2iw5_A |
666 |
199 |
45 |
2.86 |
6.67 |
14.851 |
1.522 |
T P |
| 1lrz_A |
400 |
199 |
43 |
2.80 |
11.63 |
14.778 |
1.483 |
T P |
| 2v1d_A |
666 |
199 |
42 |
2.57 |
7.14 |
14.021 |
1.572 |
T P |
| 1f5n_A |
570 |
199 |
40 |
2.70 |
5.00 |
13.703 |
1.430 |
T P |
| 1jch_A |
468 |
199 |
39 |
2.73 |
10.26 |
13.587 |
1.376 |
T P |
| 1c1g_A |
284 |
199 |
38 |
2.52 |
2.63 |
13.332 |
1.449 |
T P |
| 1deq_A |
180 |
199 |
37 |
2.45 |
8.11 |
12.627 |
1.452 |
T P |
| 2v71_A |
160 |
199 |
34 |
2.46 |
0.00 |
12.029 |
1.328 |
T P |
| 2fxo_A |
129 |
199 |
32 |
2.36 |
18.75 |
11.981 |
1.299 |
T P |
| 2efr_A |
155 |
199 |
33 |
2.62 |
6.06 |
11.920 |
1.212 |
T P |
| 2fxm_A |
126 |
199 |
32 |
2.64 |
9.38 |
11.734 |
1.167 |
T P |
| 2oqo_A |
181 |
199 |
29 |
2.59 |
3.45 |
11.213 |
1.078 |
T P |
| 1ha0_A |
494 |
199 |
29 |
2.85 |
0.00 |
10.483 |
0.983 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]