LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_401.5wLII_11277_137
Total number of 3D structures: 68
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
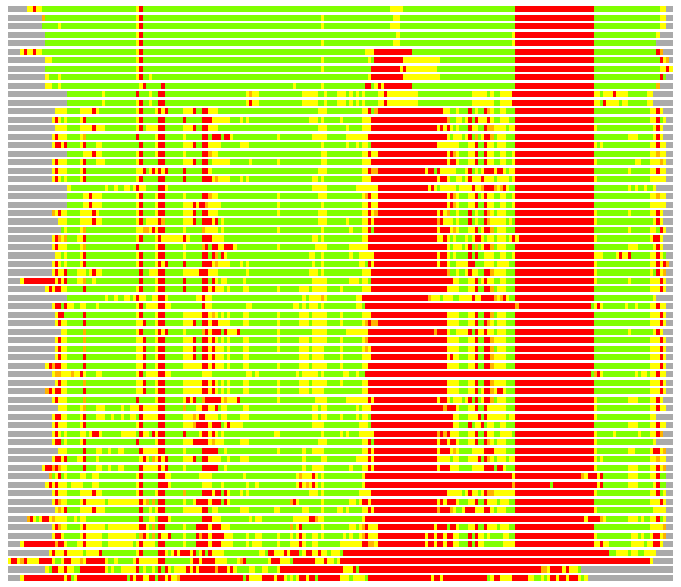
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1yg6_A |
193 |
212 |
177 |
0.83 |
17.51 |
82.322 |
19.071 |
T P |
| 1yg8_A |
179 |
212 |
173 |
0.87 |
17.92 |
80.584 |
17.849 |
T P |
| 1tg6_E |
196 |
212 |
173 |
0.95 |
16.76 |
80.000 |
16.423 |
T P |
| 2zl2_A |
174 |
212 |
170 |
0.78 |
15.29 |
79.247 |
19.256 |
T P |
| 2zl4_A |
173 |
212 |
169 |
0.82 |
14.79 |
78.668 |
18.341 |
T P |
| 1y7o_B |
178 |
212 |
164 |
1.15 |
20.73 |
75.867 |
13.084 |
T P |
| 2c8t_A |
171 |
212 |
163 |
1.29 |
15.95 |
73.827 |
11.706 |
T P |
| 2cby_D |
171 |
212 |
163 |
1.27 |
16.56 |
73.755 |
11.934 |
T P |
| 2ce3_E |
169 |
212 |
161 |
1.27 |
16.15 |
72.900 |
11.744 |
T P |
| 2f6i_C |
183 |
212 |
157 |
1.22 |
10.19 |
71.332 |
11.882 |
T P |
| 3bez_D |
479 |
212 |
156 |
1.83 |
18.59 |
66.259 |
8.085 |
T P |
| 3bf0_C |
478 |
212 |
155 |
1.85 |
19.35 |
63.633 |
7.934 |
T P |
| 2iex_A |
253 |
212 |
137 |
1.81 |
13.87 |
59.016 |
7.183 |
T P |
| 2gtr_A |
258 |
212 |
134 |
1.82 |
18.66 |
57.091 |
6.994 |
T P |
| 1q52_B |
279 |
212 |
136 |
1.91 |
12.50 |
56.902 |
6.755 |
T P |
| 2hw5_A |
260 |
212 |
130 |
1.79 |
15.38 |
56.288 |
6.864 |
T P |
| 1xx4_A |
254 |
212 |
137 |
1.89 |
13.14 |
56.225 |
6.885 |
T P |
| 1nzy_B |
269 |
212 |
134 |
1.86 |
17.91 |
55.809 |
6.841 |
T P |
| 2ej5_B |
251 |
212 |
135 |
1.84 |
15.56 |
55.538 |
6.959 |
T P |
| 1ef8_A |
256 |
212 |
136 |
1.76 |
10.29 |
54.664 |
7.308 |
T P |
| 2fbm_A |
251 |
212 |
137 |
1.88 |
15.33 |
53.271 |
6.902 |
T P |
| 3bpp_A |
216 |
212 |
138 |
1.88 |
23.91 |
53.199 |
6.967 |
T P |
| 1nzy_A |
269 |
212 |
134 |
1.85 |
17.16 |
52.985 |
6.859 |
T P |
| 1jxz_B |
269 |
212 |
133 |
1.92 |
16.54 |
52.125 |
6.572 |
T P |
| 2uzf_A |
260 |
212 |
138 |
1.86 |
13.77 |
52.083 |
7.040 |
T P |
| 1rjm_B |
240 |
212 |
135 |
1.93 |
12.59 |
51.831 |
6.650 |
T P |
| 2vx2_A |
256 |
212 |
137 |
1.94 |
13.87 |
51.812 |
6.703 |
T P |
| 2a7k_C |
238 |
212 |
135 |
1.95 |
14.07 |
51.683 |
6.573 |
T P |
| 1mj3_A |
258 |
212 |
131 |
1.87 |
15.27 |
51.286 |
6.664 |
T P |
| 2np9_A |
423 |
212 |
135 |
1.89 |
13.33 |
51.281 |
6.769 |
T P |
| 2fw2_F |
260 |
212 |
136 |
1.87 |
16.18 |
51.253 |
6.921 |
T P |
| 3fdu_A |
248 |
212 |
134 |
1.93 |
9.70 |
51.202 |
6.604 |
T P |
| 2ppy_A |
264 |
212 |
133 |
1.76 |
11.28 |
50.544 |
7.153 |
T P |
| 2vsu_C |
251 |
212 |
134 |
1.92 |
14.18 |
50.113 |
6.627 |
T P |
| 2deo_B |
200 |
212 |
132 |
1.97 |
22.73 |
49.920 |
6.364 |
T P |
| 1on3_E |
521 |
212 |
116 |
1.80 |
10.34 |
49.880 |
6.097 |
T P |
| 2vsu_F |
245 |
212 |
134 |
1.96 |
11.94 |
49.807 |
6.521 |
T P |
| 2j5i_I |
245 |
212 |
134 |
2.01 |
13.43 |
49.673 |
6.344 |
T P |
| 1ey3_A |
258 |
212 |
130 |
1.89 |
16.15 |
49.671 |
6.534 |
T P |
| 2j5i_D |
247 |
212 |
133 |
1.95 |
13.53 |
49.473 |
6.503 |
T P |
| 2j5i_A |
247 |
212 |
134 |
1.97 |
13.43 |
49.414 |
6.459 |
T P |
| 2vss_E |
240 |
212 |
132 |
1.92 |
13.64 |
49.337 |
6.550 |
T P |
| 2vsu_B |
247 |
212 |
134 |
2.00 |
12.69 |
49.273 |
6.378 |
T P |
| 1vrg_A |
515 |
212 |
118 |
1.93 |
8.47 |
49.272 |
5.822 |
T P |
| 2vsu_E |
245 |
212 |
134 |
2.01 |
12.69 |
49.081 |
6.356 |
T P |
| 2vss_F |
246 |
212 |
132 |
1.98 |
12.88 |
48.998 |
6.354 |
T P |
| 2dub_D |
257 |
212 |
130 |
1.93 |
16.15 |
48.875 |
6.418 |
T P |
| 1wdk_B |
711 |
212 |
134 |
2.02 |
14.93 |
48.868 |
6.334 |
T P |
| 1hzd_A |
266 |
212 |
132 |
2.04 |
13.64 |
47.689 |
6.180 |
T P |
| 2pbp_A |
255 |
212 |
129 |
1.96 |
18.60 |
46.979 |
6.272 |
T P |
| 3g64_A |
275 |
212 |
134 |
2.05 |
13.43 |
46.927 |
6.228 |
T P |
| 1uiy_A |
253 |
212 |
130 |
1.78 |
15.38 |
46.505 |
6.928 |
T P |
| 1dci_A |
275 |
212 |
131 |
1.91 |
10.69 |
46.146 |
6.504 |
T P |
| 1sg4_B |
258 |
212 |
138 |
2.07 |
10.87 |
46.032 |
6.366 |
T P |
| 2vre_A |
272 |
212 |
132 |
1.98 |
9.85 |
45.964 |
6.342 |
T P |
| 2f9i_A |
309 |
212 |
119 |
1.94 |
15.97 |
45.885 |
5.836 |
T P |
| 2f9y_A |
301 |
212 |
116 |
1.90 |
16.38 |
45.463 |
5.811 |
T P |
| 2f6q_B |
252 |
212 |
133 |
1.99 |
12.78 |
45.272 |
6.361 |
T P |
| 1szo_D |
251 |
212 |
127 |
2.18 |
9.45 |
44.657 |
5.566 |
T P |
| 3bpt_A |
362 |
212 |
134 |
2.11 |
6.72 |
44.194 |
6.074 |
T P |
| 1x0u_A |
518 |
212 |
117 |
2.09 |
9.40 |
43.537 |
5.347 |
T P |
| 1o8u_A |
249 |
212 |
131 |
2.22 |
8.40 |
42.458 |
5.649 |
T P |
| 2j5g_D |
249 |
212 |
131 |
2.35 |
12.98 |
41.422 |
5.346 |
T P |
| 2j5g_B |
249 |
212 |
128 |
2.31 |
14.06 |
41.126 |
5.302 |
T P |
| 1iq8_A |
577 |
212 |
87 |
2.40 |
6.90 |
27.185 |
3.476 |
T P |
| 2scu_B |
385 |
212 |
71 |
2.49 |
12.68 |
22.848 |
2.738 |
T P |
| 3d1l_B |
259 |
212 |
62 |
2.60 |
9.68 |
18.845 |
2.300 |
T P |
| 2csu_A |
435 |
212 |
55 |
2.73 |
3.64 |
16.621 |
1.946 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]