LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_41.5wLII_10850_15
Total number of 3D structures: 43
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
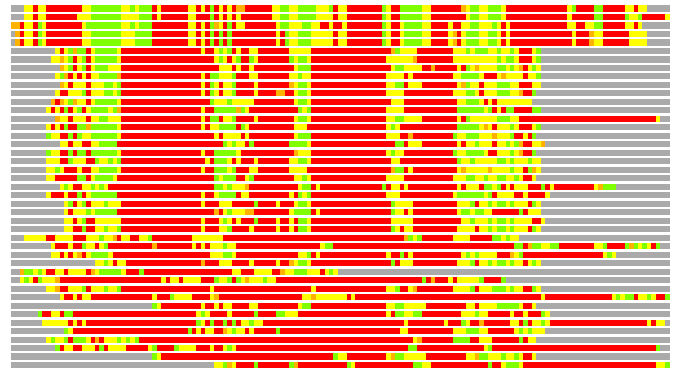
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3fbm_A |
428 |
149 |
72 |
2.48 |
4.17 |
33.800 |
2.793 |
T P |
| 2gsy_E |
433 |
149 |
70 |
2.41 |
4.29 |
33.662 |
2.786 |
T P |
| 2df7_A |
420 |
149 |
72 |
2.56 |
8.33 |
33.394 |
2.708 |
T P |
| 1wce_F |
436 |
149 |
70 |
2.56 |
7.14 |
32.839 |
2.631 |
T P |
| 1wcd_J |
421 |
149 |
70 |
2.56 |
7.14 |
32.837 |
2.631 |
T P |
| 1ii5_A |
221 |
149 |
49 |
2.62 |
10.20 |
21.603 |
1.802 |
T P |
| 1iit_A |
221 |
149 |
50 |
2.66 |
6.00 |
21.481 |
1.813 |
T P |
| 2anj_A |
262 |
149 |
46 |
2.59 |
6.52 |
20.863 |
1.711 |
T P |
| 2q2a_A |
241 |
149 |
47 |
2.76 |
8.51 |
20.645 |
1.642 |
T P |
| 1mqh_A |
260 |
149 |
46 |
2.65 |
10.87 |
20.523 |
1.670 |
T P |
| 1mqi_A |
260 |
149 |
45 |
2.68 |
13.33 |
20.320 |
1.619 |
T P |
| 1p1o_A |
260 |
149 |
46 |
2.63 |
6.52 |
20.152 |
1.683 |
T P |
| 1m5d_A |
258 |
149 |
42 |
2.33 |
4.76 |
20.074 |
1.729 |
T P |
| 3dln_A |
258 |
149 |
46 |
2.72 |
10.87 |
20.043 |
1.632 |
T P |
| 2i3v_A |
259 |
149 |
43 |
2.31 |
13.95 |
19.956 |
1.785 |
T P |
| 2v3t_B |
254 |
149 |
44 |
2.51 |
4.55 |
19.911 |
1.688 |
T P |
| 2iee_A |
251 |
149 |
42 |
2.50 |
11.90 |
19.752 |
1.616 |
T P |
| 2v3u_A |
253 |
149 |
43 |
2.31 |
6.98 |
19.689 |
1.786 |
T P |
| 1xt8_B |
251 |
149 |
46 |
2.65 |
4.35 |
19.664 |
1.676 |
T P |
| 2v25_A |
231 |
149 |
46 |
2.71 |
8.70 |
19.616 |
1.636 |
T P |
| 1ftk_A |
250 |
149 |
42 |
2.46 |
7.14 |
19.590 |
1.643 |
T P |
| 2lao_A |
238 |
149 |
44 |
2.67 |
13.64 |
19.537 |
1.588 |
T P |
| 1lbb_A |
258 |
149 |
40 |
2.41 |
7.50 |
19.398 |
1.595 |
T P |
| 1lbc_B |
260 |
149 |
43 |
2.63 |
6.98 |
19.190 |
1.576 |
T P |
| 1lb8_A |
261 |
149 |
41 |
2.66 |
7.32 |
18.369 |
1.486 |
T P |
| 3dp6_A |
258 |
149 |
43 |
2.77 |
4.65 |
18.192 |
1.500 |
T P |
| 1p1w_A |
258 |
149 |
40 |
2.69 |
5.00 |
17.775 |
1.432 |
T P |
| 2ia4_B |
278 |
149 |
42 |
2.89 |
16.67 |
17.416 |
1.406 |
T P |
| 2vha_A |
276 |
149 |
38 |
2.54 |
13.16 |
17.313 |
1.441 |
T P |
| 1lst_A |
238 |
149 |
38 |
2.64 |
10.53 |
17.205 |
1.384 |
T P |
| 2i3w_A |
258 |
149 |
38 |
2.74 |
5.26 |
16.540 |
1.337 |
T P |
| 2o1m_A |
232 |
149 |
39 |
2.67 |
2.56 |
16.447 |
1.406 |
T P |
| 3fas_A |
260 |
149 |
38 |
2.86 |
7.89 |
16.347 |
1.284 |
T P |
| 2pyy_B |
217 |
149 |
38 |
2.85 |
10.53 |
15.911 |
1.288 |
T P |
| 3b6q_A |
258 |
149 |
34 |
2.96 |
5.88 |
15.035 |
1.109 |
T P |
| 1mqd_A |
258 |
149 |
33 |
2.63 |
9.09 |
14.986 |
1.207 |
T P |
| 2uxa_A |
261 |
149 |
32 |
2.60 |
6.25 |
14.855 |
1.183 |
T P |
| 3b6w_A |
258 |
149 |
33 |
2.82 |
3.03 |
14.834 |
1.129 |
T P |
| 1hsl_A |
238 |
149 |
30 |
2.51 |
10.00 |
13.823 |
1.151 |
T P |
| 1wdn_A |
223 |
149 |
30 |
2.49 |
6.67 |
13.392 |
1.158 |
T P |
| 1pb8_A |
282 |
149 |
27 |
2.70 |
3.70 |
12.926 |
0.963 |
T P |
| 2gfe_A |
260 |
149 |
28 |
2.70 |
7.14 |
12.275 |
0.999 |
T P |
| 2gfp_A |
375 |
149 |
23 |
2.47 |
8.70 |
10.649 |
0.895 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]