LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_419.5wLII_11277_198
Total number of 3D structures: 55
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
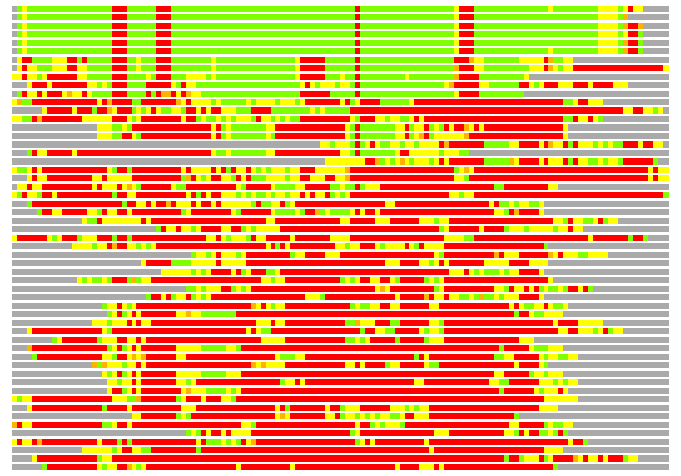
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2uy8_A |
377 |
132 |
115 |
1.02 |
15.65 |
85.769 |
10.222 |
T P |
| 1l3j_A |
372 |
132 |
113 |
0.92 |
16.81 |
85.443 |
11.027 |
T P |
| 2uya_A |
375 |
132 |
114 |
1.02 |
17.54 |
85.304 |
10.170 |
T P |
| 2uy9_A |
377 |
132 |
114 |
0.92 |
17.54 |
85.211 |
11.198 |
T P |
| 2uyb_A |
377 |
132 |
114 |
0.91 |
17.54 |
85.204 |
11.278 |
T P |
| 2v09_A |
377 |
132 |
114 |
0.93 |
16.67 |
85.045 |
11.040 |
T P |
| 1y3t_A |
330 |
132 |
91 |
1.69 |
7.69 |
64.692 |
5.097 |
T P |
| 2h0v_A |
335 |
132 |
94 |
1.79 |
7.45 |
64.601 |
4.965 |
T P |
| 1y9q_A |
178 |
132 |
81 |
1.66 |
7.41 |
57.134 |
4.604 |
T P |
| 2qnk_A |
286 |
132 |
83 |
1.92 |
6.02 |
54.608 |
4.109 |
T P |
| 2bnm_A |
194 |
132 |
78 |
1.81 |
14.10 |
50.768 |
4.074 |
T P |
| 2o0b_A |
424 |
132 |
49 |
2.50 |
6.12 |
25.023 |
1.885 |
T P |
| 2cvh_A |
220 |
132 |
45 |
2.25 |
8.89 |
24.912 |
1.914 |
T P |
| 2bjb_A |
421 |
132 |
52 |
2.57 |
1.92 |
24.893 |
1.950 |
T P |
| 2hnf_A |
130 |
132 |
41 |
2.40 |
9.76 |
24.545 |
1.641 |
T P |
| 2ho0_A |
128 |
132 |
42 |
2.28 |
7.14 |
23.770 |
1.761 |
T P |
| 2aw6_A |
287 |
132 |
47 |
2.50 |
0.00 |
22.829 |
1.806 |
T P |
| 3bdn_A |
234 |
132 |
40 |
2.30 |
10.00 |
22.562 |
1.667 |
T P |
| 2axv_B |
303 |
132 |
44 |
2.54 |
4.55 |
21.229 |
1.667 |
T P |
| 2awi_A |
298 |
132 |
41 |
2.87 |
9.76 |
20.475 |
1.379 |
T P |
| 2axu_A |
303 |
132 |
41 |
2.84 |
4.88 |
20.221 |
1.395 |
T P |
| 1f39_A |
101 |
132 |
37 |
2.50 |
0.00 |
19.280 |
1.421 |
T P |
| 2qfc_A |
284 |
132 |
41 |
2.74 |
12.20 |
19.217 |
1.444 |
T P |
| 2e0z_A |
236 |
132 |
36 |
2.65 |
11.11 |
18.441 |
1.310 |
T P |
| 1kca_A |
101 |
132 |
35 |
2.62 |
0.00 |
18.291 |
1.285 |
T P |
| 1lli_B |
92 |
132 |
36 |
2.72 |
8.33 |
17.994 |
1.276 |
T P |
| 2ef8_A |
84 |
132 |
35 |
2.73 |
8.57 |
17.286 |
1.236 |
T P |
| 2grm_A |
315 |
132 |
34 |
2.77 |
5.88 |
16.891 |
1.186 |
T P |
| 3f6w_A |
82 |
132 |
35 |
2.84 |
5.71 |
16.752 |
1.192 |
T P |
| 3clc_B |
77 |
132 |
33 |
2.82 |
9.09 |
16.210 |
1.130 |
T P |
| 2jvl_A |
107 |
132 |
32 |
2.93 |
9.38 |
15.986 |
1.055 |
T P |
| 3b7h_A |
76 |
132 |
30 |
2.45 |
13.33 |
15.875 |
1.179 |
T P |
| 1y7y_A |
69 |
132 |
30 |
2.48 |
3.33 |
15.553 |
1.164 |
T P |
| 3f52_E |
78 |
132 |
30 |
2.74 |
3.33 |
15.414 |
1.056 |
T P |
| 2p5t_E |
95 |
132 |
32 |
2.64 |
0.00 |
15.284 |
1.170 |
T P |
| 1x57_A |
91 |
132 |
32 |
2.69 |
0.00 |
15.159 |
1.148 |
T P |
| 1sq8_A |
64 |
132 |
25 |
2.41 |
8.00 |
15.111 |
0.994 |
T P |
| 1b0n_A |
103 |
132 |
32 |
2.87 |
12.50 |
15.108 |
1.078 |
T P |
| 1rio_A |
97 |
132 |
27 |
2.49 |
0.00 |
15.089 |
1.043 |
T P |
| 3cec_A |
91 |
132 |
27 |
2.83 |
11.11 |
14.730 |
0.922 |
T P |
| 1r63_A |
63 |
132 |
27 |
2.55 |
7.41 |
14.631 |
1.020 |
T P |
| 2ewt_A |
71 |
132 |
30 |
2.68 |
10.00 |
14.585 |
1.078 |
T P |
| 2r63_A |
63 |
132 |
29 |
3.01 |
3.45 |
14.545 |
0.933 |
T P |
| 1r69_A |
63 |
132 |
28 |
2.92 |
10.71 |
13.869 |
0.927 |
T P |
| 3eus_A |
85 |
132 |
28 |
2.85 |
0.00 |
13.537 |
0.950 |
T P |
| 1utx_A |
66 |
132 |
23 |
2.69 |
13.04 |
13.452 |
0.826 |
T P |
| 2b5a_A |
77 |
132 |
23 |
2.71 |
8.70 |
13.387 |
0.819 |
T P |
| 1adr_A |
76 |
132 |
25 |
2.76 |
0.00 |
13.231 |
0.873 |
T P |
| 2cro_A |
65 |
132 |
27 |
2.63 |
11.11 |
13.085 |
0.988 |
T P |
| 3dnv_B |
71 |
132 |
26 |
2.77 |
3.85 |
12.635 |
0.905 |
T P |
| 3bs3_A |
62 |
132 |
23 |
2.55 |
8.70 |
12.544 |
0.868 |
T P |
| 2axz_A |
306 |
132 |
23 |
2.63 |
8.70 |
12.332 |
0.843 |
T P |
| 1lmb_4 |
92 |
132 |
18 |
2.87 |
16.67 |
10.160 |
0.606 |
T P |
| 2k9q_A |
77 |
132 |
19 |
2.72 |
5.26 |
10.087 |
0.674 |
T P |
| 2r1j_L |
66 |
132 |
17 |
2.96 |
5.88 |
9.154 |
0.556 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]