LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_421.5wLII_11277_202
Total number of 3D structures: 66
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
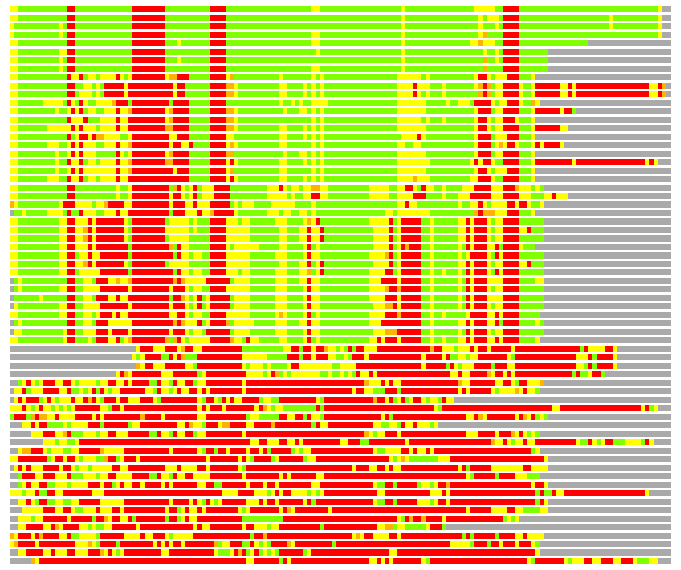
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1k6m_A |
432 |
162 |
142 |
1.09 |
22.54 |
85.115 |
11.902 |
T P |
| 2bif_A |
432 |
162 |
142 |
1.10 |
20.42 |
84.844 |
11.795 |
T P |
| 1bif_A |
432 |
162 |
142 |
1.09 |
20.42 |
84.803 |
11.925 |
T P |
| 3bif_A |
432 |
162 |
142 |
1.18 |
20.42 |
84.527 |
11.078 |
T P |
| 2axn_A |
451 |
162 |
124 |
1.33 |
21.77 |
72.620 |
8.647 |
T P |
| 1fbt_A |
190 |
162 |
114 |
1.20 |
23.68 |
67.357 |
8.800 |
T P |
| 1tip_A |
190 |
162 |
114 |
1.23 |
23.68 |
67.178 |
8.570 |
T P |
| 1c80_A |
191 |
162 |
114 |
1.28 |
23.68 |
66.485 |
8.243 |
T P |
| 3ezn_A |
236 |
162 |
106 |
1.93 |
9.43 |
56.768 |
5.217 |
T P |
| 2h4x_A |
255 |
162 |
105 |
1.93 |
6.67 |
55.673 |
5.172 |
T P |
| 2hhj_A |
254 |
162 |
105 |
1.92 |
6.67 |
55.551 |
5.202 |
T P |
| 1fzt_A |
211 |
162 |
101 |
1.91 |
7.92 |
53.795 |
5.014 |
T P |
| 1qhf_A |
240 |
162 |
105 |
1.94 |
8.57 |
52.484 |
5.153 |
T P |
| 3fdz_B |
228 |
162 |
104 |
1.95 |
9.62 |
51.898 |
5.074 |
T P |
| 1rii_A |
243 |
162 |
105 |
2.02 |
11.43 |
51.852 |
4.946 |
T P |
| 3d8h_B |
235 |
162 |
102 |
1.92 |
6.86 |
51.823 |
5.057 |
T P |
| 1yfk_A |
243 |
162 |
107 |
2.05 |
11.21 |
51.817 |
4.969 |
T P |
| 5pgm_D |
235 |
162 |
103 |
1.91 |
8.74 |
51.705 |
5.118 |
T P |
| 1e58_A |
246 |
162 |
105 |
2.00 |
9.52 |
51.522 |
5.002 |
T P |
| 1xq9_A |
241 |
162 |
104 |
2.03 |
11.54 |
51.305 |
4.874 |
T P |
| 1e59_A |
239 |
162 |
99 |
1.89 |
10.10 |
49.438 |
4.979 |
T P |
| 1ebb_A |
202 |
162 |
103 |
2.16 |
9.71 |
47.072 |
4.560 |
T P |
| 1h2e_A |
207 |
162 |
104 |
2.11 |
10.58 |
46.841 |
4.706 |
T P |
| 2a6p_A |
193 |
162 |
100 |
2.17 |
11.00 |
45.693 |
4.397 |
T P |
| 3pgm_A |
230 |
162 |
102 |
2.21 |
7.84 |
45.433 |
4.409 |
T P |
| 2p2z_A |
171 |
162 |
94 |
2.18 |
14.89 |
41.584 |
4.124 |
T P |
| 2p77_A |
171 |
162 |
92 |
2.19 |
15.22 |
41.068 |
4.023 |
T P |
| 1v37_A |
171 |
162 |
93 |
2.15 |
15.05 |
41.020 |
4.125 |
T P |
| 2p30_A |
177 |
162 |
89 |
2.10 |
15.73 |
41.005 |
4.054 |
T P |
| 2p9y_A |
171 |
162 |
90 |
2.09 |
16.67 |
40.910 |
4.118 |
T P |
| 2ekb_A |
171 |
162 |
92 |
2.17 |
15.22 |
40.672 |
4.057 |
T P |
| 2p79_A |
171 |
162 |
90 |
2.11 |
16.67 |
40.550 |
4.075 |
T P |
| 2pa0_A |
171 |
162 |
90 |
2.23 |
15.56 |
40.025 |
3.867 |
T P |
| 2ekz_A |
171 |
162 |
89 |
2.12 |
14.61 |
39.977 |
4.001 |
T P |
| 2owd_A |
171 |
162 |
89 |
2.12 |
16.85 |
39.828 |
4.002 |
T P |
| 2p6m_A |
171 |
162 |
88 |
2.13 |
15.91 |
39.493 |
3.950 |
T P |
| 2p2y_A |
170 |
162 |
90 |
2.16 |
17.78 |
39.254 |
3.988 |
T P |
| 2p6o_A |
171 |
162 |
89 |
2.23 |
15.73 |
38.553 |
3.812 |
T P |
| 2p75_A |
171 |
162 |
88 |
2.16 |
14.77 |
38.302 |
3.897 |
T P |
| 2owe_A |
170 |
162 |
90 |
2.23 |
15.56 |
38.275 |
3.864 |
T P |
| 3e4p_A |
254 |
162 |
49 |
2.63 |
10.20 |
20.484 |
1.798 |
T P |
| 2zbb_A |
255 |
162 |
46 |
2.47 |
8.70 |
20.224 |
1.793 |
T P |
| 3by9_A |
259 |
162 |
49 |
2.64 |
6.12 |
20.210 |
1.788 |
T P |
| 3e4o_A |
254 |
162 |
50 |
2.77 |
6.00 |
19.636 |
1.742 |
T P |
| 3b8t_A |
358 |
162 |
48 |
2.95 |
4.17 |
19.558 |
1.576 |
T P |
| 1xfc_A |
366 |
162 |
50 |
2.83 |
6.00 |
19.313 |
1.706 |
T P |
| 2vd9_A |
386 |
162 |
47 |
2.80 |
4.26 |
19.310 |
1.620 |
T P |
| 3c2w_C |
482 |
162 |
43 |
2.59 |
16.28 |
18.996 |
1.598 |
T P |
| 2j66_A |
390 |
162 |
43 |
2.77 |
9.30 |
18.915 |
1.496 |
T P |
| 2odo_A |
354 |
162 |
46 |
2.76 |
2.17 |
18.697 |
1.607 |
T P |
| 3b8u_A |
358 |
162 |
45 |
2.86 |
2.22 |
18.471 |
1.522 |
T P |
| 1w8g_A |
226 |
162 |
46 |
2.61 |
15.22 |
18.295 |
1.698 |
T P |
| 1vfs_A |
382 |
162 |
44 |
2.83 |
9.09 |
18.029 |
1.503 |
T P |
| 1xql_A |
381 |
162 |
41 |
2.64 |
4.88 |
17.930 |
1.495 |
T P |
| 2vd8_A |
386 |
162 |
48 |
3.09 |
6.25 |
17.796 |
1.504 |
T P |
| 2rjg_A |
358 |
162 |
44 |
2.71 |
4.55 |
17.664 |
1.565 |
T P |
| 2sfp_A |
378 |
162 |
43 |
2.76 |
6.98 |
17.614 |
1.503 |
T P |
| 2a3v_B |
320 |
162 |
43 |
2.79 |
4.65 |
17.545 |
1.485 |
T P |
| 1epv_A |
381 |
162 |
44 |
2.73 |
6.82 |
17.533 |
1.556 |
T P |
| 1bd0_A |
381 |
162 |
43 |
2.94 |
4.65 |
17.137 |
1.414 |
T P |
| 1a0p_A |
271 |
162 |
37 |
2.42 |
0.00 |
16.700 |
1.466 |
T P |
| 1rcq_A |
356 |
162 |
40 |
2.74 |
7.50 |
16.019 |
1.407 |
T P |
| 3b8w_A |
358 |
162 |
45 |
3.06 |
8.89 |
15.971 |
1.425 |
T P |
| 3b8v_A |
358 |
162 |
38 |
2.89 |
2.63 |
15.604 |
1.271 |
T P |
| 3co8_A |
360 |
162 |
35 |
2.77 |
8.57 |
14.007 |
1.219 |
T P |
| 2dy3_D |
344 |
162 |
29 |
2.84 |
3.45 |
11.755 |
0.986 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]