LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_426.5wLII_11277_212
Total number of 3D structures: 45
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
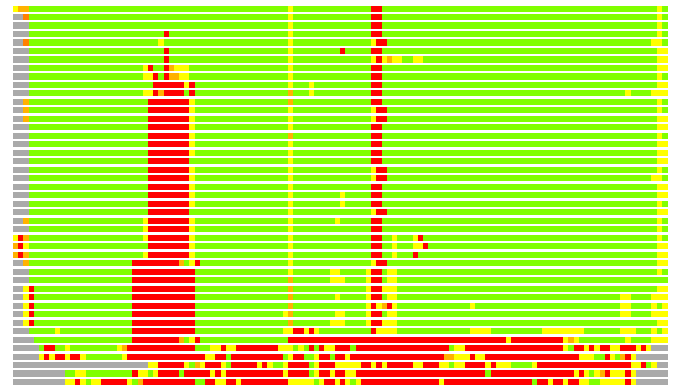
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ihg_A |
364 |
126 |
124 |
0.81 |
18.55 |
96.989 |
13.638 |
T P |
| 2gw2_A |
173 |
126 |
122 |
0.89 |
15.57 |
95.471 |
12.358 |
T P |
| 2he9_A |
172 |
126 |
121 |
0.74 |
16.53 |
95.249 |
14.335 |
T P |
| 2igv_A |
172 |
126 |
120 |
0.70 |
19.17 |
94.573 |
15.074 |
T P |
| 1a58_A |
177 |
126 |
122 |
1.05 |
14.75 |
94.552 |
10.585 |
T P |
| 2poy_B |
172 |
126 |
119 |
0.69 |
20.17 |
93.968 |
15.153 |
T P |
| 2hqj_A |
179 |
126 |
121 |
1.02 |
19.01 |
93.947 |
10.801 |
T P |
| 1qng_A |
170 |
126 |
119 |
0.98 |
18.49 |
92.955 |
10.991 |
T P |
| 1qnh_B |
170 |
126 |
119 |
1.07 |
17.65 |
92.760 |
10.192 |
T P |
| 1mzw_A |
173 |
126 |
114 |
0.82 |
17.54 |
90.121 |
12.449 |
T P |
| 1z81_A |
186 |
126 |
115 |
1.12 |
16.52 |
89.543 |
9.417 |
T P |
| 2esl_A |
181 |
126 |
114 |
0.93 |
16.67 |
89.268 |
11.056 |
T P |
| 2rmc_A |
182 |
126 |
114 |
0.93 |
16.67 |
89.244 |
11.092 |
T P |
| 1cyn_A |
178 |
126 |
114 |
0.93 |
16.67 |
89.213 |
11.074 |
T P |
| 2cfe_A |
162 |
126 |
113 |
0.71 |
18.58 |
89.146 |
13.944 |
T P |
| 1ynd_A |
164 |
126 |
113 |
0.75 |
19.47 |
89.094 |
13.357 |
T P |
| 1zcx_A |
164 |
126 |
113 |
0.72 |
21.24 |
89.084 |
13.820 |
T P |
| 2r99_A |
164 |
126 |
113 |
0.72 |
21.24 |
89.084 |
13.820 |
T P |
| 1vdn_A |
161 |
126 |
113 |
0.74 |
22.12 |
89.021 |
13.467 |
T P |
| 2z6w_A |
164 |
126 |
113 |
0.78 |
17.70 |
88.917 |
12.843 |
T P |
| 2alf_A |
164 |
126 |
113 |
0.76 |
19.47 |
88.917 |
13.092 |
T P |
| 2bit_X |
164 |
126 |
113 |
0.75 |
17.70 |
88.910 |
13.267 |
T P |
| 1zmf_A |
162 |
126 |
113 |
0.79 |
21.24 |
88.847 |
12.648 |
T P |
| 1m9e_A |
164 |
126 |
113 |
0.82 |
19.47 |
88.844 |
12.245 |
T P |
| 2cmt_A |
164 |
126 |
113 |
0.82 |
19.47 |
88.743 |
12.217 |
T P |
| 1aws_A |
164 |
126 |
114 |
1.05 |
19.30 |
88.704 |
9.924 |
T P |
| 1awq_A |
164 |
126 |
113 |
0.87 |
19.47 |
88.422 |
11.702 |
T P |
| 3bt8_A |
166 |
126 |
114 |
1.03 |
15.79 |
88.315 |
10.126 |
T P |
| 1xo7_A |
166 |
126 |
114 |
1.10 |
18.42 |
88.012 |
9.538 |
T P |
| 2haq_A |
166 |
126 |
114 |
1.06 |
16.67 |
88.008 |
9.831 |
T P |
| 1h0p_A |
182 |
126 |
112 |
1.01 |
16.96 |
87.197 |
10.118 |
T P |
| 2ok3_A |
160 |
126 |
109 |
1.06 |
18.35 |
84.440 |
9.379 |
T P |
| 2oju_B |
166 |
126 |
109 |
1.11 |
18.35 |
84.398 |
9.036 |
T P |
| 2b71_A |
169 |
126 |
109 |
1.04 |
13.76 |
84.346 |
9.532 |
T P |
| 2a2n_C |
165 |
126 |
109 |
1.17 |
20.18 |
83.636 |
8.568 |
T P |
| 2poe_A |
162 |
126 |
109 |
1.28 |
13.76 |
83.601 |
7.899 |
T P |
| 2fu0_A |
155 |
126 |
109 |
1.21 |
15.60 |
83.580 |
8.311 |
T P |
| 1zkc_A |
187 |
126 |
109 |
1.20 |
14.68 |
83.295 |
8.377 |
T P |
| 1xwn_A |
166 |
126 |
107 |
1.75 |
17.76 |
77.304 |
5.775 |
T P |
| 2c3b_A |
141 |
126 |
60 |
1.68 |
16.67 |
40.225 |
3.376 |
T P |
| 2r6g_F |
490 |
126 |
43 |
2.51 |
9.30 |
23.551 |
1.650 |
T P |
| 3fh6_F |
316 |
126 |
38 |
2.64 |
15.79 |
21.948 |
1.386 |
T P |
| 2j4w_H |
225 |
126 |
42 |
2.82 |
14.29 |
21.071 |
1.440 |
T P |
| 2onk_C |
252 |
126 |
36 |
2.54 |
5.56 |
21.050 |
1.362 |
T P |
| 2r5w_B |
345 |
126 |
39 |
2.73 |
0.00 |
21.049 |
1.380 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]