LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_438.5wLII_11277_266
Total number of 3D structures: 52
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
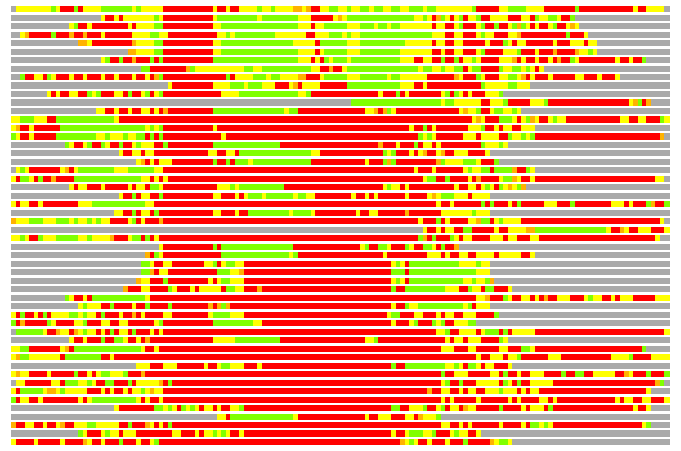
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qvr_A |
803 |
147 |
98 |
2.56 |
17.35 |
51.340 |
3.690 |
T P |
| 2dq0_A |
447 |
147 |
76 |
2.24 |
7.89 |
42.469 |
3.243 |
T P |
| 2z3y_A |
643 |
147 |
76 |
2.35 |
7.89 |
41.470 |
3.100 |
T P |
| 2hko_A |
647 |
147 |
74 |
2.45 |
8.11 |
40.080 |
2.897 |
T P |
| 2v1d_A |
666 |
147 |
73 |
2.43 |
9.59 |
39.890 |
2.887 |
T P |
| 2iw5_A |
666 |
147 |
72 |
2.43 |
9.72 |
39.697 |
2.851 |
T P |
| 2h94_A |
647 |
147 |
70 |
2.13 |
7.14 |
37.457 |
3.143 |
T P |
| 1h6k_C |
733 |
147 |
72 |
2.21 |
1.39 |
35.773 |
3.115 |
T P |
| 2dw4_A |
634 |
147 |
70 |
2.58 |
12.86 |
35.336 |
2.615 |
T P |
| 3ddh_A |
229 |
147 |
48 |
2.36 |
16.67 |
24.502 |
1.948 |
T P |
| 2w4m_A |
250 |
147 |
46 |
2.23 |
10.87 |
24.292 |
1.973 |
T P |
| 3bwl_A |
125 |
147 |
41 |
2.10 |
7.32 |
23.161 |
1.867 |
T P |
| 2gfh_A |
246 |
147 |
45 |
2.37 |
11.11 |
23.116 |
1.822 |
T P |
| 1te2_A |
218 |
147 |
43 |
2.25 |
9.30 |
22.368 |
1.830 |
T P |
| 1o08_A |
221 |
147 |
40 |
2.25 |
5.00 |
21.592 |
1.704 |
T P |
| 1lvh_A |
220 |
147 |
44 |
2.39 |
4.55 |
21.428 |
1.766 |
T P |
| 1rql_A |
257 |
147 |
40 |
2.32 |
7.50 |
20.808 |
1.652 |
T P |
| 2hoq_A |
237 |
147 |
39 |
2.37 |
7.69 |
20.776 |
1.581 |
T P |
| 2fi1_A |
187 |
147 |
40 |
2.32 |
10.00 |
20.692 |
1.652 |
T P |
| 2hsz_A |
225 |
147 |
40 |
2.18 |
2.50 |
20.650 |
1.753 |
T P |
| 2go7_C |
205 |
147 |
39 |
2.40 |
0.00 |
20.060 |
1.558 |
T P |
| 2pib_A |
216 |
147 |
42 |
2.49 |
2.38 |
19.718 |
1.623 |
T P |
| 2ioh_A |
256 |
147 |
41 |
2.48 |
4.88 |
19.599 |
1.590 |
T P |
| 2hcf_A |
225 |
147 |
38 |
2.41 |
15.79 |
19.451 |
1.515 |
T P |
| 2iof_K |
255 |
147 |
39 |
2.35 |
5.13 |
19.328 |
1.595 |
T P |
| 2iof_A |
256 |
147 |
41 |
2.73 |
0.00 |
19.217 |
1.450 |
T P |
| 3e58_A |
211 |
147 |
36 |
2.49 |
2.78 |
18.894 |
1.393 |
T P |
| 3d6j_A |
210 |
147 |
41 |
2.65 |
2.44 |
18.616 |
1.493 |
T P |
| 3eeh_A |
116 |
147 |
30 |
1.80 |
6.67 |
18.568 |
1.579 |
T P |
| 3dv9_A |
243 |
147 |
37 |
2.45 |
2.70 |
18.537 |
1.449 |
T P |
| 3f1p_B |
111 |
147 |
34 |
2.20 |
5.88 |
18.344 |
1.480 |
T P |
| 2a24_A |
107 |
147 |
32 |
1.99 |
0.00 |
18.324 |
1.534 |
T P |
| 3f1p_A |
114 |
147 |
33 |
2.22 |
3.03 |
18.233 |
1.420 |
T P |
| 2a24_B |
108 |
147 |
35 |
2.39 |
5.71 |
18.054 |
1.406 |
T P |
| 2ho4_B |
252 |
147 |
39 |
2.59 |
15.38 |
17.947 |
1.450 |
T P |
| 2r78_C |
116 |
147 |
32 |
2.46 |
6.25 |
17.890 |
1.250 |
T P |
| 1x42_A |
230 |
147 |
40 |
2.72 |
7.50 |
17.836 |
1.419 |
T P |
| 1swv_A |
257 |
147 |
35 |
2.34 |
0.00 |
17.828 |
1.434 |
T P |
| 2yy6_A |
208 |
147 |
40 |
2.67 |
5.00 |
17.674 |
1.446 |
T P |
| 2fdr_A |
222 |
147 |
34 |
2.52 |
0.00 |
17.596 |
1.296 |
T P |
| 2ah5_A |
210 |
147 |
34 |
2.20 |
0.00 |
17.561 |
1.475 |
T P |
| 2hi0_A |
240 |
147 |
39 |
2.60 |
2.56 |
17.507 |
1.444 |
T P |
| 1x0o_A |
119 |
147 |
34 |
2.48 |
2.94 |
17.505 |
1.319 |
T P |
| 2qlt_A |
251 |
147 |
38 |
2.77 |
5.26 |
17.454 |
1.323 |
T P |
| 2w43_A |
201 |
147 |
39 |
2.58 |
7.69 |
17.427 |
1.453 |
T P |
| 2nyv_A |
217 |
147 |
43 |
2.92 |
2.33 |
17.060 |
1.425 |
T P |
| 2hdo_A |
207 |
147 |
33 |
2.63 |
3.03 |
16.819 |
1.211 |
T P |
| 1rdf_A |
263 |
147 |
37 |
2.78 |
2.70 |
16.500 |
1.286 |
T P |
| 1p97_A |
114 |
147 |
28 |
2.39 |
0.00 |
15.926 |
1.126 |
T P |
| 2w11_A |
206 |
147 |
34 |
2.83 |
8.82 |
15.468 |
1.162 |
T P |
| 2b02_A |
111 |
147 |
32 |
2.54 |
3.12 |
14.758 |
1.213 |
T P |
| 1fez_A |
256 |
147 |
22 |
2.86 |
13.64 |
11.328 |
0.744 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]