LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_44.5wLII_10933_16
Total number of 3D structures: 55
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
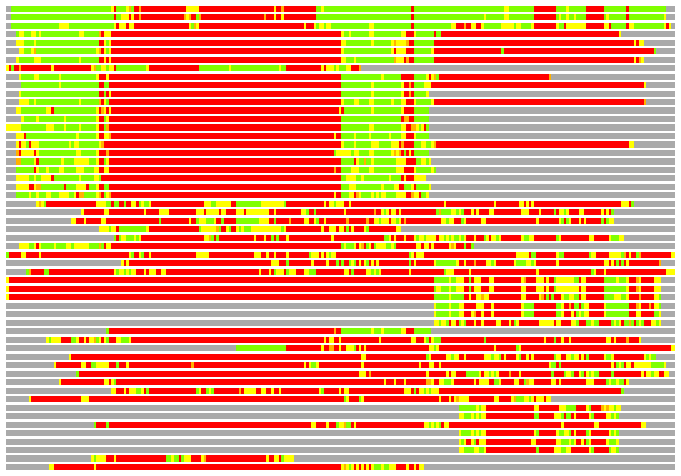
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1t5e_A |
231 |
267 |
178 |
1.28 |
22.47 |
64.456 |
12.909 |
T P |
| 1vf7_E |
252 |
267 |
178 |
1.52 |
22.47 |
64.047 |
10.972 |
T P |
| 2f1m_C |
247 |
267 |
167 |
1.87 |
22.16 |
52.735 |
8.478 |
T P |
| 1iyu_A |
79 |
267 |
74 |
1.64 |
20.27 |
25.417 |
4.253 |
T P |
| 1y8o_B |
97 |
267 |
74 |
1.95 |
18.92 |
24.843 |
3.614 |
T P |
| 2dnc_A |
98 |
267 |
71 |
1.79 |
19.72 |
24.668 |
3.757 |
T P |
| 3crk_C |
86 |
267 |
72 |
1.86 |
19.44 |
24.595 |
3.669 |
T P |
| 1wp1_A |
456 |
267 |
73 |
2.00 |
8.22 |
24.328 |
3.483 |
T P |
| 2ejm_A |
99 |
267 |
67 |
1.65 |
25.37 |
24.214 |
3.836 |
T P |
| 1z6h_A |
72 |
267 |
67 |
1.53 |
20.90 |
23.955 |
4.122 |
T P |
| 2evb_A |
74 |
267 |
66 |
1.38 |
30.30 |
23.857 |
4.452 |
T P |
| 2dne_A |
108 |
267 |
70 |
1.86 |
22.86 |
23.785 |
3.565 |
T P |
| 1bdo_A |
80 |
267 |
66 |
1.55 |
27.27 |
23.717 |
4.008 |
T P |
| 1dcz_A |
77 |
267 |
65 |
1.47 |
24.62 |
23.431 |
4.147 |
T P |
| 1a6x_A |
87 |
267 |
71 |
2.00 |
22.54 |
23.283 |
3.388 |
T P |
| 3bg3_A |
679 |
267 |
68 |
1.73 |
19.12 |
23.224 |
3.709 |
T P |
| 1qjo_A |
80 |
267 |
68 |
1.96 |
23.53 |
22.424 |
3.305 |
T P |
| 1o78_A |
84 |
267 |
67 |
2.12 |
23.88 |
21.885 |
3.020 |
T P |
| 1pmr_A |
80 |
267 |
65 |
2.04 |
20.00 |
20.390 |
3.043 |
T P |
| 1ghj_A |
79 |
267 |
69 |
1.91 |
18.84 |
20.030 |
3.440 |
T P |
| 3bdo_A |
82 |
267 |
63 |
2.00 |
26.98 |
19.939 |
2.996 |
T P |
| 1lab_A |
80 |
267 |
64 |
2.04 |
23.44 |
18.975 |
2.985 |
T P |
| 1k8m_A |
87 |
267 |
68 |
2.32 |
16.18 |
18.413 |
2.805 |
T P |
| 1cii_A |
602 |
267 |
69 |
2.42 |
11.59 |
17.795 |
2.740 |
T P |
| 2gho_D |
1196 |
267 |
72 |
2.85 |
6.94 |
17.133 |
2.437 |
T P |
| 1hqm_D |
1175 |
267 |
69 |
2.75 |
11.59 |
16.723 |
2.424 |
T P |
| 2fic_B |
201 |
267 |
62 |
2.54 |
14.52 |
16.518 |
2.346 |
T P |
| 2be5_D |
1392 |
267 |
68 |
2.54 |
4.41 |
16.484 |
2.577 |
T P |
| 1fyc_A |
106 |
267 |
64 |
2.45 |
17.19 |
15.995 |
2.506 |
T P |
| 2qf7_A |
1075 |
267 |
58 |
2.80 |
8.62 |
14.157 |
2.000 |
T P |
| 2ppl_A |
449 |
267 |
53 |
2.49 |
7.55 |
13.909 |
2.042 |
T P |
| 3bg5_A |
1137 |
267 |
56 |
2.80 |
7.14 |
13.296 |
1.929 |
T P |
| 1dpd_A |
243 |
267 |
52 |
2.59 |
9.62 |
12.915 |
1.934 |
T P |
| 1b5s_A |
242 |
267 |
52 |
2.61 |
5.77 |
12.741 |
1.920 |
T P |
| 1dpb_A |
243 |
267 |
48 |
2.49 |
10.42 |
12.713 |
1.852 |
T P |
| 1eaa_A |
243 |
267 |
46 |
2.18 |
8.70 |
12.447 |
2.016 |
T P |
| 1dpc_A |
243 |
267 |
45 |
2.21 |
8.89 |
12.433 |
1.945 |
T P |
| 1scz_A |
233 |
267 |
49 |
2.54 |
4.08 |
12.418 |
1.858 |
T P |
| 2jku_A |
35 |
267 |
35 |
1.49 |
20.00 |
12.384 |
2.198 |
T P |
| 1ddq_D |
1077 |
267 |
46 |
2.66 |
8.70 |
11.901 |
1.667 |
T P |
| 1c1g_A |
284 |
267 |
38 |
2.25 |
0.00 |
11.717 |
1.620 |
T P |
| 2ii3_A |
234 |
267 |
42 |
2.63 |
0.00 |
11.504 |
1.538 |
T P |
| 2b3y_A |
888 |
267 |
46 |
2.73 |
4.35 |
11.418 |
1.624 |
T P |
| 3b8k_A |
239 |
267 |
44 |
2.47 |
4.55 |
11.234 |
1.711 |
T P |
| 1i6v_D |
1174 |
267 |
45 |
2.82 |
6.67 |
11.070 |
1.540 |
T P |
| 1qz2_A |
285 |
267 |
43 |
2.77 |
6.98 |
10.506 |
1.497 |
T P |
| 3g5w_A |
318 |
267 |
32 |
2.88 |
9.38 |
8.128 |
1.072 |
T P |
| 1zy8_K |
44 |
267 |
30 |
2.44 |
6.67 |
7.541 |
1.179 |
T P |
| 1w4e_A |
45 |
267 |
27 |
2.33 |
7.41 |
7.206 |
1.109 |
T P |
| 2ipy_A |
847 |
267 |
26 |
2.81 |
3.85 |
7.064 |
0.895 |
T P |
| 3dva_I |
42 |
267 |
26 |
2.24 |
15.38 |
7.010 |
1.111 |
T P |
| 1ebd_C |
41 |
267 |
26 |
2.54 |
3.85 |
6.971 |
0.984 |
T P |
| 1w85_I |
42 |
267 |
25 |
2.31 |
12.00 |
6.854 |
1.035 |
T P |
| 2coo_A |
70 |
267 |
26 |
2.69 |
11.54 |
6.694 |
0.931 |
T P |
| 1zwv_A |
52 |
267 |
25 |
2.82 |
4.00 |
6.507 |
0.855 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]