LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_441.5wLII_11277_279
Total number of 3D structures: 50
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
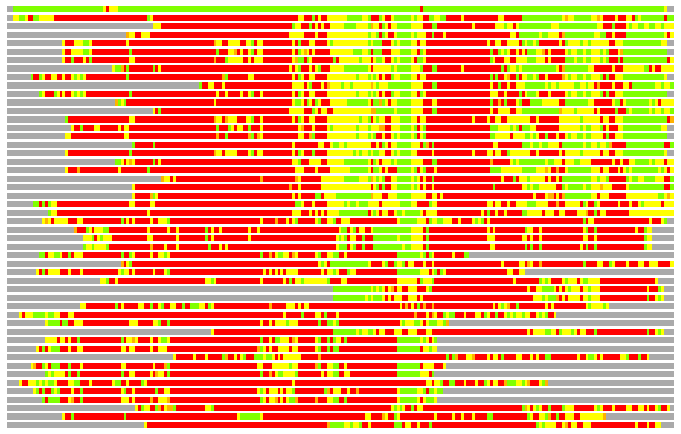
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1vqr_A |
284 |
229 |
223 |
0.53 |
18.39 |
96.989 |
35.336 |
T P |
| 2huo_A |
258 |
229 |
98 |
2.20 |
16.33 |
31.153 |
4.258 |
T P |
| 2dqb_C |
364 |
229 |
95 |
2.23 |
16.84 |
29.774 |
4.085 |
T P |
| 2pq7_A |
174 |
229 |
92 |
2.33 |
18.48 |
29.326 |
3.789 |
T P |
| 1tbf_A |
326 |
229 |
101 |
2.56 |
11.88 |
28.738 |
3.801 |
T P |
| 3bjc_A |
311 |
229 |
98 |
2.39 |
10.20 |
28.549 |
3.929 |
T P |
| 1xoz_A |
326 |
229 |
97 |
2.39 |
13.40 |
28.509 |
3.900 |
T P |
| 2o6i_A |
436 |
229 |
92 |
2.16 |
19.57 |
28.288 |
4.069 |
T P |
| 2oun_B |
328 |
229 |
96 |
2.43 |
11.46 |
27.941 |
3.791 |
T P |
| 2pgs_A |
393 |
229 |
94 |
2.37 |
17.02 |
27.625 |
3.813 |
T P |
| 2ous_B |
329 |
229 |
95 |
2.40 |
13.68 |
27.592 |
3.807 |
T P |
| 2hek_A |
369 |
229 |
89 |
2.29 |
12.36 |
27.532 |
3.727 |
T P |
| 3bg2_A |
397 |
229 |
94 |
2.36 |
13.83 |
27.524 |
3.819 |
T P |
| 2yy2_A |
326 |
229 |
97 |
2.65 |
4.12 |
27.220 |
3.525 |
T P |
| 1z1l_A |
338 |
229 |
96 |
2.44 |
9.38 |
27.183 |
3.784 |
T P |
| 2o8h_A |
307 |
229 |
93 |
2.43 |
13.98 |
27.134 |
3.679 |
T P |
| 2o08_A |
187 |
229 |
81 |
2.23 |
20.99 |
27.028 |
3.479 |
T P |
| 3dyn_A |
328 |
229 |
94 |
2.58 |
3.19 |
26.812 |
3.513 |
T P |
| 2q14_C |
406 |
229 |
92 |
2.43 |
19.57 |
26.693 |
3.635 |
T P |
| 1tbm_A |
326 |
229 |
89 |
2.40 |
4.49 |
26.563 |
3.554 |
T P |
| 2ogi_B |
194 |
229 |
84 |
2.32 |
20.24 |
26.447 |
3.469 |
T P |
| 3ccg_A |
189 |
229 |
79 |
2.38 |
17.72 |
24.679 |
3.182 |
T P |
| 2qgs_A |
209 |
229 |
80 |
2.53 |
12.50 |
23.816 |
3.046 |
T P |
| 2ouv_B |
328 |
229 |
86 |
2.74 |
6.98 |
23.672 |
3.033 |
T P |
| 1xx7_A |
172 |
229 |
81 |
2.57 |
11.11 |
22.565 |
3.029 |
T P |
| 1w25_A |
454 |
229 |
61 |
2.68 |
9.84 |
18.198 |
2.195 |
T P |
| 2br1_A |
272 |
229 |
55 |
2.50 |
5.45 |
16.189 |
2.115 |
T P |
| 2hy0_A |
272 |
229 |
53 |
2.60 |
3.77 |
15.985 |
1.966 |
T P |
| 1zlt_A |
272 |
229 |
51 |
2.38 |
5.88 |
15.945 |
2.055 |
T P |
| 2jba_A |
125 |
229 |
50 |
2.36 |
6.00 |
15.207 |
2.036 |
T P |
| 1ny5_B |
385 |
229 |
48 |
2.48 |
4.17 |
15.161 |
1.861 |
T P |
| 1kgs_A |
219 |
229 |
51 |
2.62 |
9.80 |
15.083 |
1.873 |
T P |
| 2oqr_A |
226 |
229 |
55 |
2.81 |
1.82 |
14.999 |
1.887 |
T P |
| 1zh2_A |
120 |
229 |
50 |
2.51 |
6.00 |
14.700 |
1.916 |
T P |
| 1zes_A |
121 |
229 |
50 |
2.50 |
4.00 |
14.666 |
1.922 |
T P |
| 1ys7_B |
226 |
229 |
48 |
2.88 |
12.50 |
14.164 |
1.613 |
T P |
| 2qhn_A |
268 |
229 |
49 |
2.54 |
6.12 |
14.119 |
1.858 |
T P |
| 2a9r_A |
117 |
229 |
48 |
2.39 |
8.33 |
14.113 |
1.931 |
T P |
| 1xhe_B |
122 |
229 |
48 |
2.57 |
8.33 |
14.072 |
1.796 |
T P |
| 2jb9_B |
122 |
229 |
47 |
2.50 |
6.38 |
13.895 |
1.806 |
T P |
| 1mvo_A |
121 |
229 |
45 |
2.57 |
6.67 |
13.720 |
1.686 |
T P |
| 1yvr_A |
520 |
229 |
50 |
2.83 |
6.00 |
13.686 |
1.708 |
T P |
| 2iyn_B |
124 |
229 |
45 |
2.53 |
8.89 |
13.685 |
1.713 |
T P |
| 2jba_B |
121 |
229 |
46 |
2.41 |
4.35 |
13.665 |
1.834 |
T P |
| 1ia8_A |
272 |
229 |
48 |
2.71 |
8.33 |
13.423 |
1.706 |
T P |
| 1xhf_B |
122 |
229 |
46 |
2.45 |
4.35 |
13.341 |
1.807 |
T P |
| 2a9o_A |
117 |
229 |
46 |
2.66 |
4.35 |
13.337 |
1.668 |
T P |
| 1dc7_A |
124 |
229 |
44 |
2.73 |
4.55 |
13.292 |
1.553 |
T P |
| 2gwr_A |
225 |
229 |
43 |
2.68 |
9.30 |
12.947 |
1.547 |
T P |
| 1b00_A |
122 |
229 |
39 |
2.72 |
7.69 |
11.752 |
1.384 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]