LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_442.5wLII_11277_281
Total number of 3D structures: 59
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
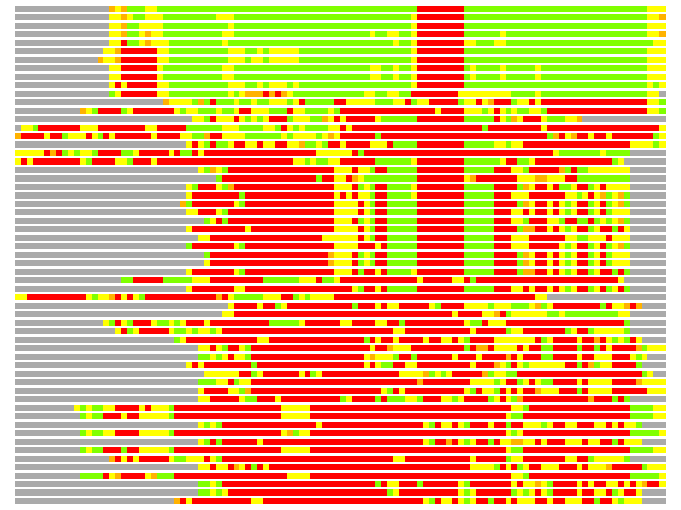
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1zfj_A |
476 |
110 |
86 |
1.09 |
23.26 |
76.934 |
7.208 |
T P |
| 1eep_A |
314 |
110 |
86 |
1.46 |
20.93 |
74.031 |
5.530 |
T P |
| 1jr1_A |
436 |
110 |
86 |
1.59 |
18.60 |
72.947 |
5.074 |
T P |
| 1nf7_A |
454 |
110 |
86 |
1.67 |
17.44 |
72.624 |
4.848 |
T P |
| 1jcn_A |
395 |
110 |
85 |
1.67 |
20.00 |
70.909 |
4.792 |
T P |
| 2c6q_B |
329 |
110 |
81 |
1.52 |
12.35 |
69.860 |
4.988 |
T P |
| 2ble_A |
337 |
110 |
82 |
1.69 |
13.41 |
69.665 |
4.580 |
T P |
| 1pvn_A |
362 |
110 |
80 |
1.43 |
12.50 |
68.337 |
5.232 |
T P |
| 1mwf_A |
362 |
110 |
80 |
1.43 |
12.50 |
68.337 |
5.233 |
T P |
| 2a7r_A |
338 |
110 |
80 |
1.42 |
12.50 |
68.118 |
5.276 |
T P |
| 1ypf_A |
295 |
110 |
77 |
2.07 |
12.99 |
61.597 |
3.549 |
T P |
| 2zg6_B |
203 |
110 |
52 |
2.50 |
11.54 |
33.286 |
1.999 |
T P |
| 1f7u_A |
606 |
110 |
47 |
2.47 |
6.38 |
28.724 |
1.829 |
T P |
| 2yvz_A |
248 |
110 |
43 |
2.44 |
9.30 |
27.757 |
1.691 |
T P |
| 2yzq_A |
224 |
110 |
37 |
2.21 |
10.81 |
26.376 |
1.604 |
T P |
| 2yvx_A |
442 |
110 |
44 |
2.79 |
11.36 |
26.226 |
1.523 |
T P |
| 2yvy_A |
248 |
110 |
41 |
2.45 |
2.44 |
25.873 |
1.611 |
T P |
| 3f1e_X |
362 |
110 |
39 |
2.38 |
5.13 |
25.351 |
1.570 |
T P |
| 2ef7_A |
127 |
110 |
35 |
2.21 |
8.57 |
24.592 |
1.516 |
T P |
| 1ey7_A |
135 |
110 |
35 |
2.51 |
2.86 |
22.606 |
1.341 |
T P |
| 3c1e_A |
131 |
110 |
34 |
2.39 |
5.88 |
22.178 |
1.365 |
T P |
| 1ez6_A |
135 |
110 |
34 |
2.49 |
0.00 |
21.872 |
1.313 |
T P |
| 2ey2_A |
136 |
110 |
30 |
2.32 |
0.00 |
21.729 |
1.239 |
T P |
| 2pzu_A |
130 |
110 |
33 |
2.51 |
0.00 |
21.666 |
1.263 |
T P |
| 2pyk_A |
130 |
110 |
33 |
2.40 |
0.00 |
21.593 |
1.321 |
T P |
| 3dmu_A |
126 |
110 |
33 |
2.28 |
0.00 |
21.586 |
1.388 |
T P |
| 1u9r_A |
129 |
110 |
29 |
2.31 |
0.00 |
21.211 |
1.202 |
T P |
| 2f0t_A |
135 |
110 |
33 |
2.54 |
3.03 |
21.192 |
1.249 |
T P |
| 2oxp_A |
129 |
110 |
30 |
2.27 |
0.00 |
21.038 |
1.264 |
T P |
| 1ey9_A |
135 |
110 |
31 |
2.36 |
0.00 |
20.819 |
1.259 |
T P |
| 1eyc_A |
135 |
110 |
31 |
2.44 |
3.23 |
20.676 |
1.222 |
T P |
| 2pw5_A |
130 |
110 |
29 |
2.32 |
0.00 |
20.379 |
1.198 |
T P |
| 1nsn_S |
138 |
110 |
27 |
2.13 |
7.41 |
19.866 |
1.210 |
T P |
| 3eji_A |
130 |
110 |
28 |
2.39 |
0.00 |
19.688 |
1.123 |
T P |
| 2sns_A |
141 |
110 |
29 |
2.59 |
3.45 |
19.265 |
1.076 |
T P |
| 1eya_A |
135 |
110 |
30 |
2.77 |
0.00 |
18.728 |
1.046 |
T P |
| 1f2m_A |
136 |
110 |
27 |
2.56 |
3.70 |
18.631 |
1.016 |
T P |
| 1kdc_A |
136 |
110 |
29 |
2.32 |
13.79 |
18.330 |
1.197 |
T P |
| 2rks_A |
135 |
110 |
27 |
2.43 |
11.11 |
17.679 |
1.066 |
T P |
| 3d6c_A |
135 |
110 |
28 |
2.89 |
10.71 |
17.485 |
0.936 |
T P |
| 2f0w_A |
136 |
110 |
29 |
2.93 |
3.45 |
17.261 |
0.956 |
T P |
| 2rdf_A |
125 |
110 |
28 |
2.79 |
7.14 |
17.163 |
0.970 |
T P |
| 2f0s_A |
135 |
110 |
27 |
2.61 |
11.11 |
16.977 |
0.996 |
T P |
| 2rbm_A |
130 |
110 |
28 |
2.76 |
0.00 |
16.896 |
0.980 |
T P |
| 1kda_A |
136 |
110 |
30 |
2.89 |
3.33 |
16.725 |
1.004 |
T P |
| 1kdb_A |
136 |
110 |
27 |
3.01 |
11.11 |
16.670 |
0.868 |
T P |
| 1nuc_A |
135 |
110 |
27 |
2.46 |
7.41 |
16.517 |
1.053 |
T P |
| 3d8g_A |
130 |
110 |
26 |
2.55 |
3.85 |
16.416 |
0.982 |
T P |
| 3ero_A |
129 |
110 |
25 |
2.44 |
4.00 |
16.385 |
0.983 |
T P |
| 3erq_A |
129 |
110 |
26 |
2.70 |
15.38 |
16.300 |
0.929 |
T P |
| 1syb_A |
137 |
110 |
26 |
2.49 |
7.69 |
16.285 |
1.006 |
T P |
| 3bdc_A |
129 |
110 |
27 |
2.88 |
3.70 |
16.210 |
0.907 |
T P |
| 3evq_A |
129 |
110 |
25 |
2.45 |
4.00 |
16.050 |
0.980 |
T P |
| 1ey8_A |
135 |
110 |
26 |
2.63 |
7.69 |
15.813 |
0.951 |
T P |
| 2qdb_A |
125 |
110 |
28 |
3.06 |
14.29 |
15.700 |
0.886 |
T P |
| 3d4d_A |
129 |
110 |
24 |
2.54 |
4.17 |
15.552 |
0.910 |
T P |
| 2exz_A |
135 |
110 |
24 |
2.69 |
0.00 |
15.229 |
0.861 |
T P |
| 2f0p_A |
135 |
110 |
22 |
2.49 |
4.55 |
14.855 |
0.851 |
T P |
| 2f0d_A |
136 |
110 |
26 |
3.04 |
7.69 |
14.554 |
0.828 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]