LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_445.5wLII_11277_291
Total number of 3D structures: 63
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
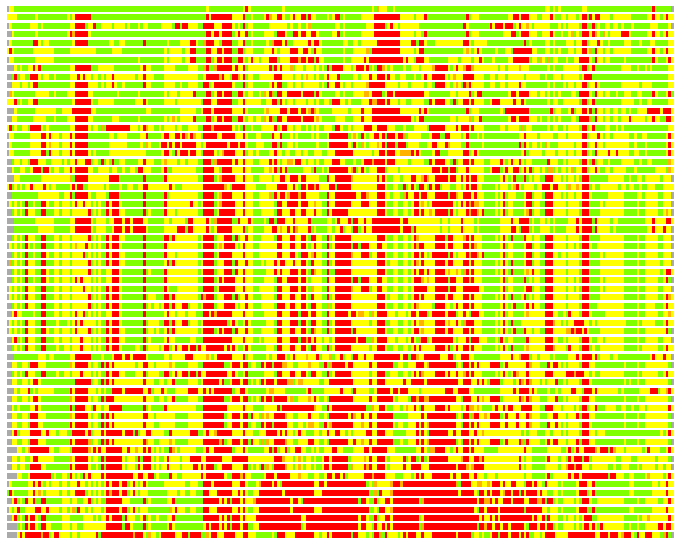
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1sbp_A |
309 |
254 |
250 |
1.31 |
20.00 |
93.609 |
17.781 |
T P |
| 2h5y_B |
233 |
254 |
204 |
2.02 |
14.22 |
62.888 |
9.612 |
T P |
| 2qry_C |
318 |
254 |
217 |
2.09 |
12.90 |
59.788 |
9.891 |
T P |
| 1amf_A |
231 |
254 |
199 |
2.01 |
15.08 |
59.695 |
9.435 |
T P |
| 3c9h_B |
339 |
254 |
209 |
2.07 |
10.05 |
57.817 |
9.629 |
T P |
| 3cg3_A |
315 |
254 |
209 |
2.23 |
13.88 |
56.550 |
8.976 |
T P |
| 3cg1_A |
293 |
254 |
198 |
2.13 |
14.14 |
53.376 |
8.891 |
T P |
| 1mrp_A |
309 |
254 |
208 |
2.31 |
9.62 |
53.153 |
8.648 |
T P |
| 1y4t_A |
317 |
254 |
212 |
2.42 |
9.43 |
52.252 |
8.401 |
T P |
| 1d9y_A |
309 |
254 |
211 |
2.32 |
7.58 |
51.984 |
8.736 |
T P |
| 2ons_A |
314 |
254 |
192 |
2.16 |
15.10 |
51.756 |
8.486 |
T P |
| 1xvx_A |
311 |
254 |
206 |
2.31 |
9.22 |
51.379 |
8.539 |
T P |
| 1atg_A |
231 |
254 |
186 |
2.26 |
13.44 |
51.234 |
7.898 |
T P |
| 3cij_A |
291 |
254 |
191 |
2.23 |
14.66 |
50.633 |
8.192 |
T P |
| 1q35_A |
317 |
254 |
197 |
2.27 |
11.68 |
50.118 |
8.302 |
T P |
| 1poy_1 |
323 |
254 |
197 |
2.31 |
13.71 |
49.899 |
8.164 |
T P |
| 1pot_A |
322 |
254 |
194 |
2.31 |
12.89 |
49.048 |
8.063 |
T P |
| 1a99_A |
341 |
254 |
200 |
2.48 |
10.50 |
48.634 |
7.744 |
T P |
| 1xvy_A |
307 |
254 |
199 |
2.46 |
9.55 |
47.741 |
7.764 |
T P |
| 2v84_A |
319 |
254 |
194 |
2.44 |
10.82 |
47.635 |
7.644 |
T P |
| 1r6z_Z |
499 |
254 |
190 |
2.50 |
8.42 |
47.208 |
7.318 |
T P |
| 1eu8_A |
407 |
254 |
193 |
2.57 |
7.77 |
46.938 |
7.230 |
T P |
| 3eht_A |
452 |
254 |
183 |
2.43 |
7.65 |
46.860 |
7.238 |
T P |
| 3d4g_E |
472 |
254 |
186 |
2.44 |
7.53 |
46.689 |
7.315 |
T P |
| 1mh3_A |
421 |
254 |
192 |
2.48 |
8.33 |
46.535 |
7.431 |
T P |
| 3fj7_A |
231 |
254 |
185 |
2.38 |
14.59 |
46.429 |
7.465 |
T P |
| 3fjm_A |
231 |
254 |
183 |
2.35 |
16.39 |
46.348 |
7.484 |
T P |
| 3c4m_A |
469 |
254 |
194 |
2.48 |
8.25 |
46.296 |
7.527 |
T P |
| 3ehs_A |
457 |
254 |
191 |
2.43 |
7.33 |
46.254 |
7.560 |
T P |
| 2vgq_A |
461 |
254 |
187 |
2.42 |
7.49 |
46.169 |
7.429 |
T P |
| 1hsj_A |
487 |
254 |
194 |
2.49 |
8.25 |
46.109 |
7.496 |
T P |
| 1mg1_A |
450 |
254 |
195 |
2.51 |
8.72 |
46.078 |
7.469 |
T P |
| 3ehu_A |
441 |
254 |
187 |
2.37 |
7.49 |
46.024 |
7.586 |
T P |
| 1mdp_1 |
363 |
254 |
185 |
2.41 |
7.57 |
45.865 |
7.369 |
T P |
| 1y4c_A |
480 |
254 |
193 |
2.51 |
8.29 |
45.696 |
7.396 |
T P |
| 1a7l_A |
380 |
254 |
191 |
2.51 |
8.90 |
45.532 |
7.307 |
T P |
| 2ok2_A |
402 |
254 |
185 |
2.46 |
8.65 |
45.305 |
7.234 |
T P |
| 1mdq_A |
371 |
254 |
189 |
2.51 |
8.47 |
45.179 |
7.238 |
T P |
| 1nmu_A |
368 |
254 |
190 |
2.54 |
7.89 |
45.007 |
7.204 |
T P |
| 1t0k_A |
365 |
254 |
181 |
2.41 |
8.29 |
44.689 |
7.221 |
T P |
| 3f5f_A |
656 |
254 |
186 |
2.52 |
9.68 |
44.469 |
7.106 |
T P |
| 3fir_A |
231 |
254 |
178 |
2.38 |
14.04 |
44.026 |
7.172 |
T P |
| 1qw0_A |
308 |
254 |
175 |
2.58 |
8.57 |
43.944 |
6.531 |
T P |
| 3dm0_A |
675 |
254 |
184 |
2.58 |
8.70 |
43.703 |
6.857 |
T P |
| 2o68_A |
307 |
254 |
172 |
2.51 |
10.47 |
43.642 |
6.586 |
T P |
| 2hxw_B |
230 |
254 |
180 |
2.51 |
14.44 |
43.580 |
6.895 |
T P |
| 1qvs_A |
308 |
254 |
172 |
2.56 |
9.30 |
43.469 |
6.469 |
T P |
| 1elj_A |
380 |
254 |
176 |
2.46 |
11.36 |
43.419 |
6.867 |
T P |
| 2o6a_A |
307 |
254 |
174 |
2.60 |
8.62 |
43.156 |
6.452 |
T P |
| 2o69_A |
308 |
254 |
173 |
2.59 |
9.25 |
43.114 |
6.438 |
T P |
| 2pt1_A |
317 |
254 |
182 |
2.55 |
15.93 |
42.962 |
6.860 |
T P |
| 1nnf_A |
308 |
254 |
174 |
2.59 |
10.34 |
42.871 |
6.471 |
T P |
| 2voz_B |
315 |
254 |
179 |
2.57 |
11.73 |
42.701 |
6.699 |
T P |
| 1xc1_A |
309 |
254 |
177 |
2.67 |
7.34 |
41.864 |
6.384 |
T P |
| 1o7t_A |
309 |
254 |
172 |
2.65 |
8.14 |
41.273 |
6.254 |
T P |
| 1y9u_A |
314 |
254 |
160 |
2.51 |
13.12 |
39.428 |
6.123 |
T P |
| 2hq0_A |
388 |
254 |
154 |
2.56 |
11.04 |
38.652 |
5.783 |
T P |
| 2heu_A |
391 |
254 |
141 |
2.44 |
8.51 |
36.568 |
5.555 |
T P |
| 1n3x_A |
366 |
254 |
134 |
2.35 |
8.96 |
35.232 |
5.463 |
T P |
| 2hfb_A |
383 |
254 |
132 |
2.44 |
9.85 |
34.064 |
5.188 |
T P |
| 3csg_A |
458 |
254 |
131 |
2.47 |
6.87 |
33.661 |
5.093 |
T P |
| 2nvu_B |
789 |
254 |
125 |
2.38 |
6.40 |
32.889 |
5.037 |
T P |
| 3csb_A |
464 |
254 |
123 |
2.73 |
11.38 |
29.849 |
4.347 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]