LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_451.5wLII_11284_3
Total number of 3D structures: 61
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
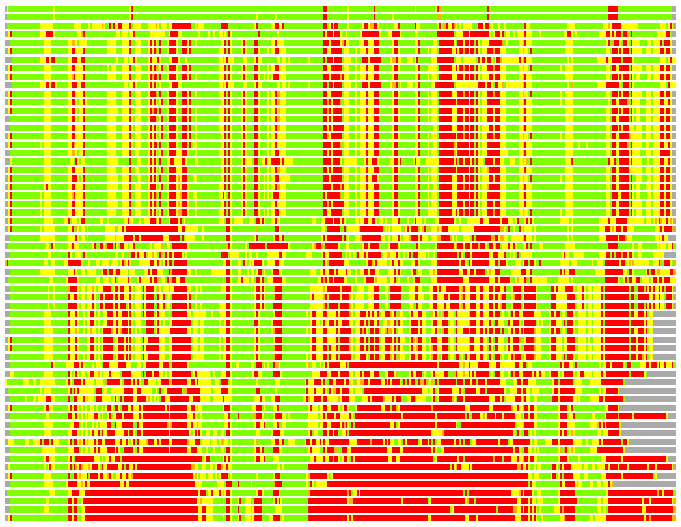
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3c96_A |
381 |
360 |
346 |
0.64 |
25.43 |
95.542 |
46.761 |
T P |
| 2rgj_A |
376 |
360 |
347 |
0.79 |
25.07 |
95.379 |
39.043 |
T P |
| 2vou_A |
393 |
360 |
309 |
1.97 |
20.39 |
67.441 |
14.936 |
T P |
| 2dkh_A |
614 |
360 |
283 |
1.83 |
21.20 |
60.705 |
14.643 |
T P |
| 1ykj_A |
394 |
360 |
285 |
1.93 |
18.60 |
58.532 |
14.040 |
T P |
| 1bf3_A |
391 |
360 |
284 |
1.95 |
17.96 |
57.694 |
13.836 |
T P |
| 1pn0_C |
656 |
360 |
290 |
1.99 |
20.34 |
57.688 |
13.882 |
T P |
| 1bgn_A |
391 |
360 |
284 |
1.97 |
17.96 |
57.599 |
13.725 |
T P |
| 1cj4_A |
392 |
360 |
283 |
1.96 |
17.67 |
57.457 |
13.764 |
T P |
| 1foh_C |
656 |
360 |
289 |
2.00 |
20.07 |
57.424 |
13.756 |
T P |
| 1bkw_A |
391 |
360 |
284 |
1.96 |
18.66 |
56.447 |
13.761 |
T P |
| 1bgj_A |
391 |
360 |
284 |
1.98 |
19.01 |
56.404 |
13.682 |
T P |
| 1cc4_A |
391 |
360 |
284 |
1.94 |
18.31 |
56.396 |
13.902 |
T P |
| 1pbd_A |
391 |
360 |
284 |
1.97 |
18.31 |
56.132 |
13.712 |
T P |
| 1pbf_A |
391 |
360 |
286 |
1.99 |
17.83 |
56.112 |
13.704 |
T P |
| 1pxc_A |
394 |
360 |
284 |
1.99 |
18.66 |
56.060 |
13.558 |
T P |
| 1pxa_A |
394 |
360 |
284 |
1.98 |
17.96 |
55.955 |
13.639 |
T P |
| 1cj2_A |
391 |
360 |
283 |
1.96 |
18.02 |
55.907 |
13.715 |
T P |
| 1k0i_A |
394 |
360 |
282 |
2.08 |
18.79 |
55.880 |
12.931 |
T P |
| 1doc_A |
394 |
360 |
284 |
1.98 |
18.31 |
55.802 |
13.643 |
T P |
| 1dob_A |
394 |
360 |
284 |
1.99 |
17.96 |
55.796 |
13.560 |
T P |
| 1pbe_A |
391 |
360 |
283 |
1.99 |
18.02 |
55.502 |
13.564 |
T P |
| 1cc6_A |
391 |
360 |
283 |
1.98 |
17.67 |
55.446 |
13.635 |
T P |
| 1pxb_A |
394 |
360 |
285 |
1.99 |
18.25 |
55.056 |
13.632 |
T P |
| 1cj3_A |
392 |
360 |
285 |
2.01 |
17.89 |
54.803 |
13.504 |
T P |
| 3e1t_A |
437 |
360 |
292 |
2.13 |
19.18 |
54.568 |
13.113 |
T P |
| 2qa2_A |
489 |
360 |
262 |
2.16 |
22.90 |
54.217 |
11.610 |
T P |
| 2qa1_A |
488 |
360 |
267 |
2.14 |
21.35 |
51.794 |
11.919 |
T P |
| 3c4a_A |
365 |
360 |
260 |
1.89 |
16.92 |
51.504 |
13.054 |
T P |
| 2r0c_A |
509 |
360 |
269 |
2.14 |
20.45 |
51.002 |
12.023 |
T P |
| 2e4g_A |
528 |
360 |
264 |
2.19 |
14.02 |
48.766 |
11.507 |
T P |
| 3cgv_A |
382 |
360 |
265 |
2.27 |
17.74 |
48.224 |
11.160 |
T P |
| 2pyx_A |
526 |
360 |
260 |
2.22 |
13.85 |
47.804 |
11.211 |
T P |
| 1x31_B |
402 |
360 |
232 |
2.25 |
12.07 |
42.951 |
9.873 |
T P |
| 2gah_B |
403 |
360 |
228 |
2.21 |
11.40 |
42.855 |
9.865 |
T P |
| 2gag_B |
403 |
360 |
227 |
2.18 |
12.33 |
42.624 |
9.950 |
T P |
| 1l9c_A |
385 |
360 |
239 |
2.40 |
13.81 |
42.496 |
9.565 |
T P |
| 1zov_A |
381 |
360 |
232 |
2.30 |
14.22 |
42.417 |
9.670 |
T P |
| 2gf3_A |
385 |
360 |
235 |
2.39 |
14.47 |
42.235 |
9.424 |
T P |
| 3bhk_A |
385 |
360 |
231 |
2.33 |
14.29 |
42.201 |
9.522 |
T P |
| 1b3m_A |
385 |
360 |
230 |
2.32 |
14.35 |
42.014 |
9.504 |
T P |
| 1el9_A |
385 |
360 |
228 |
2.27 |
14.91 |
42.000 |
9.621 |
T P |
| 2gmh_A |
581 |
360 |
219 |
2.20 |
17.35 |
41.798 |
9.524 |
T P |
| 2oln_A |
385 |
360 |
222 |
2.43 |
15.77 |
39.374 |
8.788 |
T P |
| 2iid_A |
483 |
360 |
204 |
2.43 |
16.18 |
38.028 |
8.053 |
T P |
| 2ivd_A |
449 |
360 |
205 |
2.32 |
12.20 |
37.662 |
8.455 |
T P |
| 2bxr_A |
445 |
360 |
209 |
2.45 |
14.35 |
37.053 |
8.199 |
T P |
| 1knr_A |
529 |
360 |
186 |
2.10 |
15.05 |
36.907 |
8.443 |
T P |
| 2e5v_A |
472 |
360 |
183 |
2.11 |
13.66 |
35.889 |
8.274 |
T P |
| 1qo8_A |
564 |
360 |
182 |
2.07 |
14.84 |
35.505 |
8.390 |
T P |
| 2gqf_A |
401 |
360 |
179 |
2.07 |
17.32 |
35.035 |
8.235 |
T P |
| 1b37_B |
462 |
360 |
185 |
2.48 |
13.51 |
34.187 |
7.182 |
T P |
| 2i0z_A |
416 |
360 |
176 |
2.23 |
14.77 |
34.001 |
7.560 |
T P |
| 1chu_A |
478 |
360 |
163 |
2.04 |
17.18 |
33.044 |
7.617 |
T P |
| 2vq7_C |
445 |
360 |
163 |
2.30 |
10.43 |
31.334 |
6.793 |
T P |
| 1w4x_A |
533 |
360 |
149 |
2.16 |
14.09 |
30.151 |
6.593 |
T P |
| 2ywl_A |
180 |
360 |
141 |
1.97 |
23.40 |
29.551 |
6.813 |
T P |
| 1q1r_A |
421 |
360 |
135 |
1.86 |
21.48 |
28.242 |
6.874 |
T P |
| 2a8x_A |
464 |
360 |
134 |
2.08 |
14.18 |
26.913 |
6.154 |
T P |
| 1ebd_A |
455 |
360 |
131 |
1.93 |
11.45 |
26.702 |
6.439 |
T P |
| 2eq6_A |
460 |
360 |
131 |
2.13 |
18.32 |
26.050 |
5.866 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]