LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_452.5wLII_11284_4
Total number of 3D structures: 38
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
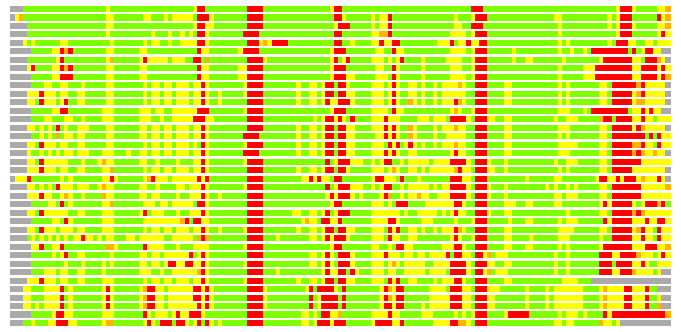
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2obw_A |
252 |
159 |
142 |
0.84 |
16.90 |
87.802 |
15.095 |
T P |
| 2p18_A |
283 |
159 |
141 |
1.50 |
12.06 |
84.067 |
8.801 |
T P |
| 1xm8_A |
254 |
159 |
139 |
1.28 |
14.39 |
83.309 |
10.069 |
T P |
| 1qh5_A |
260 |
159 |
139 |
1.45 |
10.07 |
82.313 |
8.946 |
T P |
| 2gcu_A |
244 |
159 |
135 |
1.85 |
12.59 |
75.840 |
6.909 |
T P |
| 1sml_A |
266 |
159 |
126 |
1.71 |
12.70 |
72.694 |
6.946 |
T P |
| 1vme_A |
402 |
159 |
130 |
1.95 |
5.38 |
72.134 |
6.354 |
T P |
| 2qjs_A |
248 |
159 |
126 |
1.79 |
13.49 |
71.949 |
6.671 |
T P |
| 2qin_C |
265 |
159 |
125 |
1.78 |
14.40 |
71.733 |
6.658 |
T P |
| 3fcz_A |
219 |
159 |
133 |
2.04 |
10.53 |
70.470 |
6.214 |
T P |
| 2uyx_A |
218 |
159 |
134 |
2.11 |
11.19 |
69.825 |
6.074 |
T P |
| 1bmc_A |
213 |
159 |
133 |
2.13 |
9.77 |
69.438 |
5.973 |
T P |
| 2gmn_A |
264 |
159 |
125 |
1.83 |
6.40 |
69.147 |
6.477 |
T P |
| 1ddk_A |
220 |
159 |
128 |
2.02 |
10.16 |
68.737 |
6.032 |
T P |
| 2nyp_A |
215 |
159 |
133 |
2.14 |
9.77 |
68.609 |
5.934 |
T P |
| 2nzf_A |
215 |
159 |
130 |
2.04 |
9.23 |
68.469 |
6.063 |
T P |
| 2bc2_A |
218 |
159 |
132 |
2.07 |
9.85 |
67.638 |
6.089 |
T P |
| 1mqo_A |
221 |
159 |
133 |
2.15 |
9.77 |
67.533 |
5.905 |
T P |
| 1ko3_A |
230 |
159 |
129 |
2.06 |
10.85 |
66.452 |
5.963 |
T P |
| 2nze_B |
217 |
159 |
135 |
2.18 |
8.89 |
66.132 |
5.925 |
T P |
| 1p9e_A |
294 |
159 |
128 |
2.02 |
10.94 |
65.429 |
6.045 |
T P |
| 1ko2_A |
230 |
159 |
129 |
2.11 |
10.85 |
65.103 |
5.830 |
T P |
| 2yz3_A |
225 |
159 |
130 |
2.11 |
10.00 |
64.814 |
5.877 |
T P |
| 2ohh_A |
403 |
159 |
127 |
1.95 |
8.66 |
64.766 |
6.207 |
T P |
| 1m2x_A |
219 |
159 |
134 |
2.19 |
11.19 |
64.259 |
5.850 |
T P |
| 2cfu_A |
627 |
159 |
129 |
2.06 |
6.98 |
63.264 |
5.976 |
T P |
| 2bfl_B |
220 |
159 |
132 |
2.11 |
9.85 |
63.188 |
5.973 |
T P |
| 1dxk_A |
221 |
159 |
133 |
2.16 |
9.02 |
62.947 |
5.881 |
T P |
| 1ycg_A |
398 |
159 |
128 |
1.96 |
3.91 |
62.160 |
6.223 |
T P |
| 1jje_B |
221 |
159 |
129 |
2.07 |
11.63 |
61.449 |
5.955 |
T P |
| 1jjt_A |
220 |
159 |
126 |
2.01 |
10.32 |
60.604 |
5.984 |
T P |
| 1wup_A |
217 |
159 |
131 |
2.13 |
9.92 |
60.201 |
5.877 |
T P |
| 2bg7_B |
219 |
159 |
119 |
2.00 |
10.92 |
59.437 |
5.660 |
T P |
| 2br6_A |
240 |
159 |
124 |
2.08 |
8.06 |
58.480 |
5.676 |
T P |
| 3dha_A |
254 |
159 |
125 |
2.11 |
8.00 |
58.450 |
5.654 |
T P |
| 2a7m_A |
250 |
159 |
125 |
2.09 |
7.20 |
58.346 |
5.706 |
T P |
| 2cg3_A |
627 |
159 |
118 |
1.97 |
9.32 |
55.982 |
5.696 |
T P |
| 1ztc_A |
209 |
159 |
113 |
1.97 |
10.62 |
55.355 |
5.468 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]