LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_460.5wLII_11346_17
Total number of 3D structures: 56
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
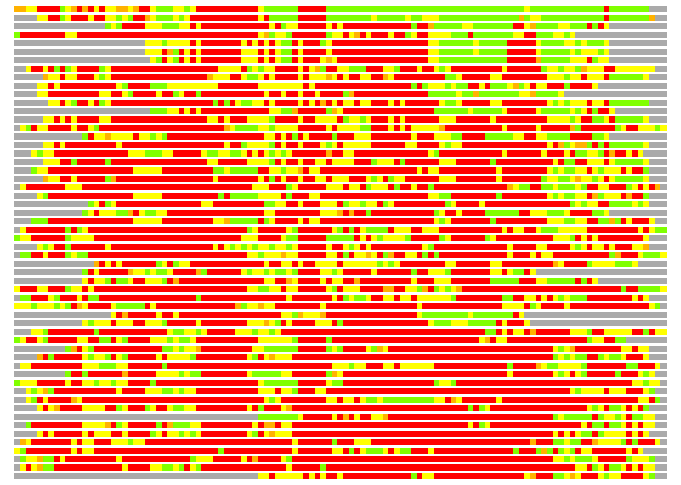
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1v7w_A |
779 |
115 |
86 |
1.93 |
9.30 |
66.387 |
4.245 |
T P |
| 2cqs_A |
822 |
115 |
81 |
1.93 |
8.64 |
59.847 |
3.987 |
T P |
| 1xdp_A |
687 |
115 |
47 |
2.21 |
4.26 |
30.743 |
2.035 |
T P |
| 2p24_A |
182 |
115 |
41 |
2.42 |
2.44 |
29.082 |
1.626 |
T P |
| 1v0w_A |
496 |
115 |
45 |
2.36 |
6.67 |
28.157 |
1.830 |
T P |
| 1f0i_A |
496 |
115 |
43 |
2.34 |
6.98 |
27.860 |
1.764 |
T P |
| 2ze9_A |
504 |
115 |
43 |
2.31 |
9.30 |
27.702 |
1.782 |
T P |
| 3pgm_A |
230 |
115 |
51 |
2.74 |
13.73 |
27.279 |
1.794 |
T P |
| 1e59_A |
239 |
115 |
49 |
2.85 |
6.12 |
27.255 |
1.661 |
T P |
| 2ze4_A |
501 |
115 |
47 |
2.68 |
6.38 |
27.109 |
1.690 |
T P |
| 1vqz_A |
330 |
115 |
40 |
2.28 |
10.00 |
26.979 |
1.684 |
T P |
| 1e58_A |
246 |
115 |
47 |
2.71 |
8.51 |
26.909 |
1.675 |
T P |
| 1byr_A |
152 |
115 |
39 |
2.30 |
12.82 |
26.328 |
1.622 |
T P |
| 1rii_A |
243 |
115 |
45 |
2.69 |
4.44 |
26.053 |
1.610 |
T P |
| 3fdz_B |
228 |
115 |
46 |
2.80 |
8.70 |
25.935 |
1.584 |
T P |
| 1x2g_C |
337 |
115 |
44 |
2.52 |
4.55 |
25.110 |
1.679 |
T P |
| 3d8h_B |
235 |
115 |
42 |
2.72 |
16.67 |
24.424 |
1.487 |
T P |
| 3ezn_A |
236 |
115 |
42 |
2.62 |
2.38 |
24.278 |
1.545 |
T P |
| 1yfk_A |
243 |
115 |
42 |
2.65 |
7.14 |
24.142 |
1.525 |
T P |
| 2ikq_A |
262 |
115 |
42 |
2.71 |
4.76 |
24.100 |
1.493 |
T P |
| 1qhf_A |
240 |
115 |
44 |
2.73 |
2.27 |
23.983 |
1.557 |
T P |
| 1h2e_A |
207 |
115 |
39 |
2.63 |
7.69 |
23.917 |
1.429 |
T P |
| 2h0q_A |
261 |
115 |
39 |
2.64 |
10.26 |
23.620 |
1.425 |
T P |
| 1xq9_A |
241 |
115 |
37 |
2.60 |
10.81 |
23.554 |
1.370 |
T P |
| 2ars_A |
245 |
115 |
40 |
2.67 |
2.50 |
23.499 |
1.445 |
T P |
| 3d4i_A |
273 |
115 |
38 |
2.46 |
2.63 |
23.289 |
1.486 |
T P |
| 2hhj_A |
254 |
115 |
38 |
2.52 |
0.00 |
23.189 |
1.451 |
T P |
| 1fbt_A |
190 |
115 |
38 |
2.65 |
2.63 |
23.165 |
1.381 |
T P |
| 5pgm_D |
235 |
115 |
38 |
2.77 |
5.26 |
22.911 |
1.326 |
T P |
| 2eoa_A |
171 |
115 |
40 |
2.63 |
7.50 |
22.818 |
1.463 |
T P |
| 2h4x_A |
255 |
115 |
41 |
2.93 |
9.76 |
22.780 |
1.355 |
T P |
| 1w66_A |
218 |
115 |
38 |
2.53 |
10.53 |
22.666 |
1.443 |
T P |
| 2qht_A |
210 |
115 |
38 |
2.63 |
10.53 |
22.452 |
1.390 |
T P |
| 1v37_A |
171 |
115 |
37 |
2.68 |
5.41 |
22.403 |
1.330 |
T P |
| 1fzt_A |
211 |
115 |
39 |
2.63 |
10.26 |
22.263 |
1.431 |
T P |
| 3fjy_B |
352 |
115 |
37 |
2.65 |
8.11 |
21.775 |
1.347 |
T P |
| 2c8m_B |
245 |
115 |
30 |
2.37 |
3.33 |
21.608 |
1.217 |
T P |
| 2qhs_A |
210 |
115 |
38 |
2.66 |
5.26 |
21.506 |
1.379 |
T P |
| 1k6m_A |
432 |
115 |
37 |
2.64 |
10.81 |
21.306 |
1.352 |
T P |
| 1ebb_A |
202 |
115 |
35 |
2.53 |
2.86 |
21.289 |
1.329 |
T P |
| 3bif_A |
432 |
115 |
34 |
2.56 |
2.94 |
21.067 |
1.279 |
T P |
| 2a6p_A |
193 |
115 |
34 |
2.56 |
2.94 |
20.716 |
1.279 |
T P |
| 2ekb_A |
171 |
115 |
38 |
2.68 |
5.26 |
20.645 |
1.366 |
T P |
| 1bif_A |
432 |
115 |
32 |
2.44 |
9.38 |
19.727 |
1.261 |
T P |
| 3eoz_B |
175 |
115 |
33 |
2.55 |
3.03 |
19.531 |
1.246 |
T P |
| 2p9z_A |
171 |
115 |
34 |
2.75 |
2.94 |
19.492 |
1.194 |
T P |
| 1ujc_A |
156 |
115 |
33 |
2.74 |
9.09 |
19.390 |
1.161 |
T P |
| 2owe_A |
170 |
115 |
31 |
2.82 |
6.45 |
19.044 |
1.063 |
T P |
| 3f2i_F |
167 |
115 |
30 |
2.40 |
6.67 |
18.816 |
1.199 |
T P |
| 2p9f_A |
171 |
115 |
32 |
2.75 |
0.00 |
18.815 |
1.121 |
T P |
| 2rfl_H |
156 |
115 |
34 |
3.01 |
5.88 |
18.452 |
1.092 |
T P |
| 2f5t_X |
233 |
115 |
30 |
2.82 |
3.33 |
18.151 |
1.028 |
T P |
| 1c80_A |
191 |
115 |
30 |
2.61 |
3.33 |
17.623 |
1.109 |
T P |
| 2p77_A |
171 |
115 |
29 |
2.72 |
3.45 |
16.140 |
1.027 |
T P |
| 2ekz_A |
171 |
115 |
26 |
2.43 |
0.00 |
16.025 |
1.029 |
T P |
| 2enu_A |
171 |
115 |
26 |
2.83 |
3.85 |
14.925 |
0.887 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]