LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_464.5wLII_11346_28
Total number of 3D structures: 40
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
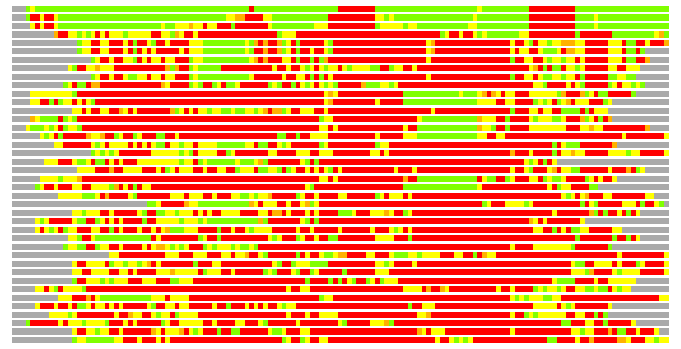
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2oqo_A |
181 |
141 |
119 |
0.65 |
16.81 |
84.067 |
15.905 |
T P |
| 2olv_B |
618 |
141 |
110 |
1.49 |
15.45 |
75.161 |
6.928 |
T P |
| 3dwk_C |
623 |
141 |
103 |
1.66 |
14.56 |
67.332 |
5.842 |
T P |
| 2gv9_B |
1035 |
141 |
55 |
2.49 |
7.27 |
27.037 |
2.124 |
T P |
| 2dxn_A |
271 |
141 |
54 |
2.71 |
3.70 |
24.728 |
1.925 |
T P |
| 3d03_A |
274 |
141 |
52 |
2.61 |
1.92 |
24.647 |
1.921 |
T P |
| 2hy1_A |
227 |
141 |
51 |
2.53 |
13.73 |
24.108 |
1.941 |
T P |
| 2dfj_A |
267 |
141 |
53 |
2.78 |
9.43 |
24.102 |
1.843 |
T P |
| 2hyo_A |
227 |
141 |
51 |
2.46 |
9.80 |
24.064 |
1.994 |
T P |
| 2qjc_A |
222 |
141 |
48 |
2.45 |
6.25 |
23.994 |
1.880 |
T P |
| 3d2l_C |
242 |
141 |
49 |
2.51 |
4.08 |
23.823 |
1.880 |
T P |
| 1wzn_A |
244 |
141 |
46 |
2.41 |
13.04 |
23.720 |
1.836 |
T P |
| 2hyp_A |
230 |
141 |
49 |
2.78 |
12.24 |
23.522 |
1.702 |
T P |
| 3g5l_A |
222 |
141 |
43 |
2.34 |
4.65 |
23.429 |
1.765 |
T P |
| 3bkw_A |
219 |
141 |
47 |
2.50 |
8.51 |
23.110 |
1.806 |
T P |
| 3ccf_B |
242 |
141 |
45 |
2.41 |
4.44 |
22.758 |
1.796 |
T P |
| 1knw_A |
421 |
141 |
47 |
2.40 |
8.51 |
22.443 |
1.882 |
T P |
| 1ii7_A |
333 |
141 |
48 |
3.09 |
12.50 |
22.220 |
1.503 |
T P |
| 2bq8_X |
304 |
141 |
46 |
2.48 |
6.52 |
22.060 |
1.782 |
T P |
| 1s8e_A |
333 |
141 |
49 |
2.79 |
6.12 |
21.875 |
1.697 |
T P |
| 1vl5_A |
231 |
141 |
42 |
2.40 |
9.52 |
21.868 |
1.680 |
T P |
| 1xxl_A |
234 |
141 |
40 |
2.38 |
10.00 |
21.627 |
1.614 |
T P |
| 1qfc_A |
287 |
141 |
48 |
2.81 |
0.00 |
21.619 |
1.650 |
T P |
| 2o57_A |
282 |
141 |
44 |
2.59 |
2.27 |
21.404 |
1.636 |
T P |
| 3f4k_A |
254 |
141 |
48 |
2.79 |
4.17 |
21.311 |
1.661 |
T P |
| 1war_A |
310 |
141 |
42 |
2.42 |
7.14 |
21.116 |
1.665 |
T P |
| 1xm7_A |
186 |
141 |
47 |
2.90 |
4.26 |
20.716 |
1.567 |
T P |
| 2yqz_A |
261 |
141 |
37 |
2.21 |
5.41 |
20.417 |
1.599 |
T P |
| 1ute_A |
302 |
141 |
41 |
2.56 |
9.76 |
20.376 |
1.540 |
T P |
| 1v73_A |
331 |
141 |
44 |
2.79 |
4.55 |
20.302 |
1.523 |
T P |
| 2gs9_A |
211 |
141 |
44 |
2.67 |
6.82 |
20.013 |
1.588 |
T P |
| 3dsd_A |
333 |
141 |
46 |
2.87 |
15.22 |
19.798 |
1.550 |
T P |
| 2zbm_A |
331 |
141 |
41 |
2.64 |
4.88 |
19.706 |
1.498 |
T P |
| 1g6q_1 |
328 |
141 |
39 |
2.73 |
2.56 |
19.434 |
1.378 |
T P |
| 3bus_A |
251 |
141 |
39 |
2.52 |
7.69 |
18.915 |
1.488 |
T P |
| 1qhw_A |
300 |
141 |
38 |
2.85 |
2.63 |
18.732 |
1.288 |
T P |
| 3e7p_A |
253 |
141 |
42 |
2.87 |
2.38 |
18.296 |
1.414 |
T P |
| 3dsc_A |
333 |
141 |
39 |
2.87 |
5.13 |
18.171 |
1.313 |
T P |
| 2z72_A |
338 |
141 |
41 |
2.93 |
7.32 |
18.018 |
1.353 |
T P |
| 3g07_A |
193 |
141 |
38 |
2.51 |
7.89 |
17.793 |
1.457 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]