LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_47.5wLII_10933_25
Total number of 3D structures: 110
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
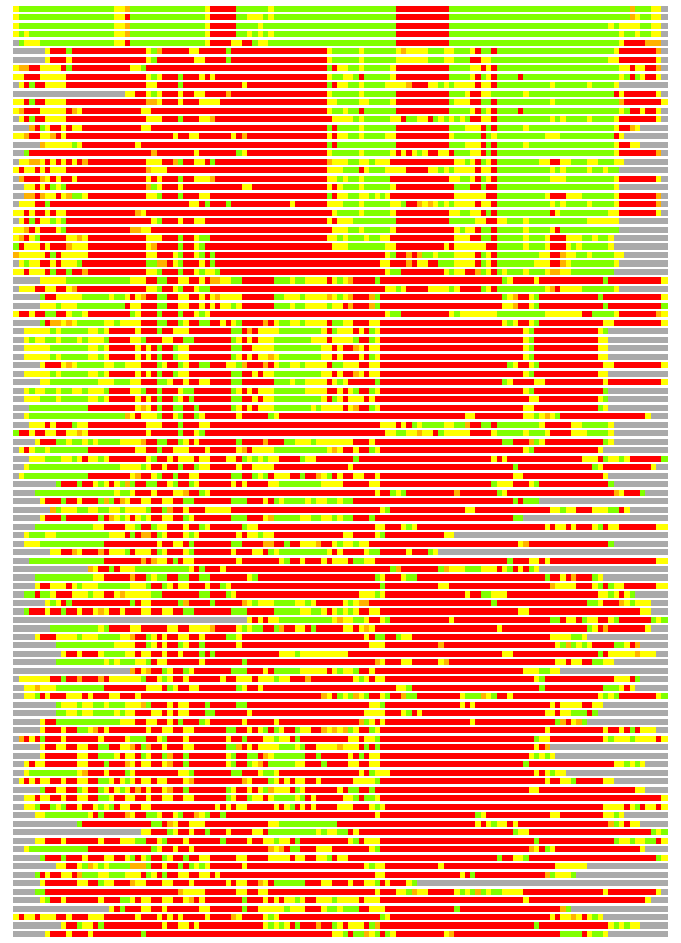
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ssq_D |
257 |
123 |
107 |
1.12 |
27.10 |
84.738 |
8.775 |
T P |
| 1t3d_A |
262 |
123 |
106 |
1.15 |
26.42 |
84.079 |
8.499 |
T P |
| 1s80_A |
241 |
123 |
107 |
1.23 |
27.10 |
83.964 |
8.060 |
T P |
| 1ssm_A |
240 |
123 |
107 |
1.32 |
27.10 |
83.490 |
7.548 |
T P |
| 3f1x_A |
276 |
123 |
100 |
1.46 |
18.00 |
79.094 |
6.417 |
T P |
| 1mr7_A |
203 |
123 |
62 |
2.01 |
4.84 |
43.426 |
2.941 |
T P |
| 1mr9_A |
203 |
123 |
61 |
1.91 |
4.92 |
43.269 |
3.042 |
T P |
| 3fs8_A |
259 |
123 |
58 |
2.20 |
15.52 |
41.391 |
2.523 |
T P |
| 1g97_A |
446 |
123 |
59 |
2.20 |
6.78 |
40.385 |
2.570 |
T P |
| 3cj8_A |
219 |
123 |
59 |
2.17 |
5.08 |
40.130 |
2.594 |
T P |
| 3bfp_A |
184 |
123 |
53 |
1.88 |
7.55 |
39.880 |
2.682 |
T P |
| 1j2z_A |
259 |
123 |
59 |
2.24 |
8.47 |
39.471 |
2.523 |
T P |
| 1hm9_A |
458 |
123 |
56 |
2.23 |
7.14 |
39.468 |
2.407 |
T P |
| 1kgq_A |
274 |
123 |
63 |
2.35 |
3.17 |
39.341 |
2.573 |
T P |
| 2npo_A |
193 |
123 |
52 |
1.94 |
3.85 |
38.886 |
2.543 |
T P |
| 1hv9_B |
450 |
123 |
55 |
2.19 |
16.36 |
38.863 |
2.400 |
T P |
| 3bsw_A |
191 |
123 |
53 |
2.01 |
3.77 |
38.729 |
2.512 |
T P |
| 1krr_A |
200 |
123 |
54 |
2.02 |
3.70 |
38.477 |
2.544 |
T P |
| 2jf2_A |
264 |
123 |
57 |
2.55 |
7.02 |
38.409 |
2.152 |
T P |
| 3eg4_A |
279 |
123 |
59 |
2.33 |
6.78 |
38.332 |
2.425 |
T P |
| 1v3w_A |
173 |
123 |
55 |
2.27 |
7.27 |
38.075 |
2.321 |
T P |
| 2qkx_A |
388 |
123 |
51 |
1.79 |
7.84 |
36.727 |
2.692 |
T P |
| 2eg0_B |
171 |
123 |
54 |
2.47 |
3.70 |
36.294 |
2.098 |
T P |
| 1tdt_A |
256 |
123 |
61 |
2.38 |
3.28 |
36.067 |
2.464 |
T P |
| 2v0h_A |
450 |
123 |
50 |
2.13 |
10.00 |
35.090 |
2.242 |
T P |
| 3d98_A |
390 |
123 |
54 |
2.16 |
9.26 |
35.042 |
2.386 |
T P |
| 1fxj_A |
327 |
123 |
50 |
2.07 |
2.00 |
34.466 |
2.302 |
T P |
| 3ect_A |
176 |
123 |
51 |
2.44 |
7.84 |
33.767 |
2.009 |
T P |
| 1ocx_A |
182 |
123 |
52 |
2.43 |
9.62 |
33.286 |
2.059 |
T P |
| 2aq9_A |
262 |
123 |
57 |
2.46 |
3.51 |
32.265 |
2.225 |
T P |
| 2qia_A |
262 |
123 |
52 |
2.46 |
3.85 |
30.831 |
2.035 |
T P |
| 2iu8_A |
346 |
123 |
49 |
2.39 |
4.08 |
29.820 |
1.969 |
T P |
| 2ch1_A |
387 |
123 |
51 |
2.35 |
3.92 |
29.382 |
2.079 |
T P |
| 1xhd_A |
172 |
123 |
49 |
2.54 |
2.04 |
29.032 |
1.854 |
T P |
| 1j04_A |
386 |
123 |
52 |
2.65 |
0.00 |
28.971 |
1.889 |
T P |
| 1h0c_A |
385 |
123 |
51 |
2.46 |
1.96 |
28.837 |
1.995 |
T P |
| 2p2o_A |
184 |
123 |
50 |
2.45 |
10.00 |
28.691 |
1.959 |
T P |
| 2rfv_A |
390 |
123 |
52 |
2.60 |
5.77 |
28.563 |
1.929 |
T P |
| 1t3i_A |
406 |
123 |
48 |
2.38 |
0.00 |
28.388 |
1.933 |
T P |
| 1elu_A |
382 |
123 |
50 |
2.35 |
10.00 |
28.287 |
2.041 |
T P |
| 1kmj_A |
405 |
123 |
48 |
2.38 |
4.17 |
27.960 |
1.932 |
T P |
| 1i29_A |
405 |
123 |
47 |
2.45 |
6.38 |
27.646 |
1.845 |
T P |
| 1y4i_A |
396 |
123 |
51 |
2.60 |
5.88 |
27.543 |
1.887 |
T P |
| 1jf9_A |
405 |
123 |
47 |
2.36 |
4.26 |
27.521 |
1.912 |
T P |
| 1vjo_A |
377 |
123 |
47 |
2.25 |
4.26 |
27.506 |
1.997 |
T P |
| 1n2t_A |
385 |
123 |
47 |
2.31 |
8.51 |
27.406 |
1.951 |
T P |
| 1c0n_A |
404 |
123 |
47 |
2.36 |
2.13 |
27.368 |
1.913 |
T P |
| 2e7j_A |
344 |
123 |
45 |
2.31 |
4.44 |
27.006 |
1.870 |
T P |
| 1qz9_A |
404 |
123 |
42 |
2.34 |
7.14 |
26.707 |
1.724 |
T P |
| 3eev_C |
208 |
123 |
47 |
2.41 |
2.13 |
26.683 |
1.873 |
T P |
| 3ftt_A |
188 |
123 |
46 |
2.62 |
2.17 |
26.167 |
1.693 |
T P |
| 1ibj_A |
380 |
123 |
46 |
2.56 |
6.52 |
25.451 |
1.730 |
T P |
| 2z9v_A |
392 |
123 |
44 |
2.37 |
6.82 |
25.360 |
1.778 |
T P |
| 2dr1_A |
381 |
123 |
46 |
2.66 |
8.70 |
24.655 |
1.668 |
T P |
| 2hdy_A |
403 |
123 |
39 |
2.26 |
10.26 |
24.062 |
1.650 |
T P |
| 1b9i_A |
384 |
123 |
39 |
2.38 |
7.69 |
23.877 |
1.573 |
T P |
| 2huf_A |
384 |
123 |
40 |
2.37 |
7.50 |
23.674 |
1.620 |
T P |
| 1fje_B |
175 |
123 |
35 |
2.31 |
2.86 |
23.211 |
1.453 |
T P |
| 2cqc_A |
95 |
123 |
37 |
2.14 |
0.00 |
23.171 |
1.652 |
T P |
| 1x4b_A |
116 |
123 |
42 |
2.84 |
0.00 |
22.934 |
1.429 |
T P |
| 2dnz_A |
95 |
123 |
37 |
2.27 |
5.41 |
22.931 |
1.562 |
T P |
| 1fxl_A |
167 |
123 |
40 |
2.48 |
5.00 |
22.825 |
1.549 |
T P |
| 1p3w_B |
385 |
123 |
39 |
2.36 |
7.69 |
22.719 |
1.586 |
T P |
| 1ecx_B |
365 |
123 |
38 |
2.41 |
2.63 |
22.689 |
1.515 |
T P |
| 2jis_B |
487 |
123 |
38 |
2.50 |
7.89 |
22.618 |
1.463 |
T P |
| 1eg5_B |
365 |
123 |
39 |
2.49 |
2.56 |
22.579 |
1.503 |
T P |
| 3bn1_A |
367 |
123 |
37 |
2.41 |
13.51 |
22.491 |
1.475 |
T P |
| 1whw_A |
99 |
123 |
34 |
2.05 |
5.88 |
22.437 |
1.581 |
T P |
| 2cjk_A |
167 |
123 |
41 |
2.60 |
2.44 |
22.427 |
1.517 |
T P |
| 2yrr_A |
352 |
123 |
41 |
2.56 |
7.32 |
22.044 |
1.541 |
T P |
| 1lw4_D |
343 |
123 |
39 |
2.64 |
0.00 |
22.043 |
1.425 |
T P |
| 1x5t_A |
96 |
123 |
38 |
2.45 |
0.00 |
21.982 |
1.492 |
T P |
| 3dr7_A |
367 |
123 |
36 |
2.29 |
2.78 |
21.842 |
1.504 |
T P |
| 3f9t_A |
394 |
123 |
40 |
2.76 |
2.50 |
21.782 |
1.401 |
T P |
| 1hd0_A |
75 |
123 |
38 |
2.65 |
5.26 |
21.422 |
1.382 |
T P |
| 1b7f_A |
167 |
123 |
37 |
2.50 |
2.70 |
21.276 |
1.423 |
T P |
| 2dis_A |
109 |
123 |
40 |
2.76 |
10.00 |
21.165 |
1.399 |
T P |
| 2fy1_A |
108 |
123 |
36 |
2.51 |
2.78 |
21.093 |
1.377 |
T P |
| 2d9p_A |
103 |
123 |
34 |
2.51 |
2.94 |
20.978 |
1.304 |
T P |
| 1cvj_A |
169 |
123 |
42 |
2.85 |
7.14 |
20.962 |
1.424 |
T P |
| 2dgo_A |
115 |
123 |
34 |
2.31 |
2.94 |
20.873 |
1.412 |
T P |
| 3cai_A |
396 |
123 |
38 |
2.65 |
7.89 |
20.831 |
1.380 |
T P |
| 1d9a_A |
85 |
123 |
36 |
2.64 |
0.00 |
20.828 |
1.313 |
T P |
| 1uaw_A |
77 |
123 |
38 |
2.61 |
0.00 |
20.805 |
1.402 |
T P |
| 1wtb_A |
79 |
123 |
34 |
2.47 |
0.00 |
20.705 |
1.322 |
T P |
| 1po6_A |
183 |
123 |
38 |
2.82 |
5.26 |
20.571 |
1.299 |
T P |
| 1pgz_A |
183 |
123 |
39 |
2.62 |
0.00 |
20.553 |
1.432 |
T P |
| 2err_A |
88 |
123 |
40 |
2.73 |
12.50 |
20.480 |
1.411 |
T P |
| 1p1t_A |
104 |
123 |
36 |
2.49 |
13.89 |
20.459 |
1.390 |
T P |
| 3sxl_A |
157 |
123 |
37 |
2.60 |
5.41 |
20.321 |
1.372 |
T P |
| 1m32_B |
362 |
123 |
34 |
2.45 |
2.94 |
20.157 |
1.332 |
T P |
| 1fnx_H |
174 |
123 |
38 |
2.84 |
10.53 |
19.894 |
1.292 |
T P |
| 2cq3_A |
103 |
123 |
34 |
2.61 |
2.94 |
19.798 |
1.254 |
T P |
| 2dh7_A |
105 |
123 |
37 |
2.69 |
2.70 |
19.324 |
1.324 |
T P |
| 1up1_A |
170 |
123 |
39 |
2.92 |
10.26 |
19.212 |
1.292 |
T P |
| 2jrs_A |
108 |
123 |
32 |
2.38 |
0.00 |
19.107 |
1.291 |
T P |
| 2cpd_A |
99 |
123 |
32 |
2.52 |
6.25 |
19.072 |
1.223 |
T P |
| 2dgu_A |
103 |
123 |
31 |
2.37 |
3.23 |
18.908 |
1.254 |
T P |
| 1l3k_A |
163 |
123 |
35 |
2.68 |
5.71 |
18.683 |
1.258 |
T P |
| 3dr4_C |
368 |
123 |
31 |
2.52 |
6.45 |
18.616 |
1.184 |
T P |
| 2dnm_A |
103 |
123 |
33 |
2.70 |
0.00 |
18.591 |
1.180 |
T P |
| 1iqt_A |
75 |
123 |
34 |
2.79 |
0.00 |
18.532 |
1.176 |
T P |
| 2mss_A |
75 |
123 |
34 |
2.67 |
8.82 |
18.468 |
1.225 |
T P |
| 1x5s_A |
102 |
123 |
32 |
2.67 |
0.00 |
18.141 |
1.156 |
T P |
| 2qfj_B |
194 |
123 |
33 |
2.65 |
6.06 |
18.114 |
1.201 |
T P |
| 2dh8_A |
105 |
123 |
34 |
2.88 |
17.65 |
17.697 |
1.141 |
T P |
| 2dk2_A |
97 |
123 |
30 |
2.75 |
3.33 |
16.363 |
1.052 |
T P |
| 1b9h_A |
384 |
123 |
30 |
2.97 |
6.67 |
16.252 |
0.977 |
T P |
| 1ha1_A |
167 |
123 |
30 |
2.71 |
3.33 |
16.038 |
1.068 |
T P |
| 2dgs_A |
99 |
123 |
24 |
2.46 |
0.00 |
14.351 |
0.939 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]