LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_470.5wLII_11346_37
Total number of 3D structures: 47
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
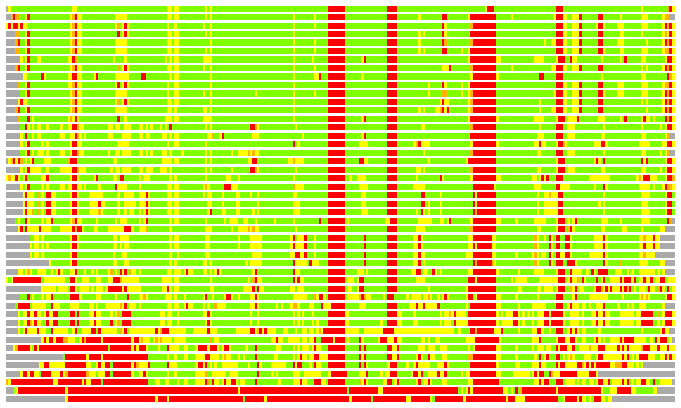
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1vyh_C |
310 |
282 |
263 |
0.83 |
12.93 |
91.808 |
28.145 |
T P |
| 2g99_A |
304 |
282 |
245 |
1.48 |
10.61 |
82.179 |
15.513 |
T P |
| 2g9a_A |
310 |
282 |
246 |
1.49 |
10.57 |
81.960 |
15.498 |
T P |
| 2h6n_B |
305 |
282 |
244 |
1.43 |
10.25 |
81.893 |
15.972 |
T P |
| 2co0_A |
304 |
282 |
245 |
1.46 |
10.61 |
81.829 |
15.727 |
T P |
| 2h9m_A |
304 |
282 |
245 |
1.47 |
10.61 |
81.822 |
15.582 |
T P |
| 3dm0_A |
675 |
282 |
244 |
1.43 |
11.48 |
81.556 |
15.904 |
T P |
| 2h9l_A |
321 |
282 |
244 |
1.46 |
10.25 |
81.549 |
15.628 |
T P |
| 1erj_C |
357 |
282 |
241 |
1.35 |
8.71 |
81.512 |
16.596 |
T P |
| 3emh_A |
300 |
282 |
243 |
1.42 |
10.29 |
81.503 |
16.012 |
T P |
| 2h14_A |
303 |
282 |
245 |
1.48 |
10.61 |
81.431 |
15.507 |
T P |
| 2gnq_A |
316 |
282 |
244 |
1.46 |
10.25 |
81.392 |
15.614 |
T P |
| 2cnx_A |
306 |
282 |
244 |
1.50 |
10.66 |
81.299 |
15.216 |
T P |
| 3frx_B |
313 |
282 |
243 |
1.51 |
10.70 |
81.078 |
15.094 |
T P |
| 1tbg_A |
340 |
282 |
244 |
1.59 |
10.66 |
80.839 |
14.447 |
T P |
| 2pbi_D |
354 |
282 |
245 |
1.58 |
8.98 |
80.762 |
14.602 |
T P |
| 3fm0_A |
328 |
282 |
243 |
1.57 |
7.41 |
80.647 |
14.593 |
T P |
| 1got_B |
339 |
282 |
247 |
1.67 |
10.93 |
80.504 |
13.922 |
T P |
| 1p22_A |
402 |
282 |
239 |
1.51 |
10.88 |
80.027 |
14.814 |
T P |
| 2bcj_B |
339 |
282 |
245 |
1.69 |
10.61 |
79.756 |
13.688 |
T P |
| 1gxr_A |
335 |
282 |
242 |
1.65 |
12.40 |
79.426 |
13.819 |
T P |
| 2hes_X |
308 |
282 |
245 |
1.70 |
7.76 |
79.109 |
13.602 |
T P |
| 3cfv_B |
393 |
282 |
243 |
1.72 |
7.41 |
78.470 |
13.382 |
T P |
| 3cfs_B |
383 |
282 |
242 |
1.73 |
7.44 |
78.263 |
13.239 |
T P |
| 3c99_A |
380 |
282 |
243 |
1.73 |
7.41 |
78.204 |
13.308 |
T P |
| 1l0q_A |
391 |
282 |
246 |
1.76 |
14.23 |
77.289 |
13.234 |
T P |
| 1a0r_B |
339 |
282 |
239 |
1.89 |
10.46 |
77.066 |
12.026 |
T P |
| 2pm7_D |
288 |
282 |
234 |
1.66 |
5.98 |
76.459 |
13.332 |
T P |
| 2pm6_D |
288 |
282 |
234 |
1.67 |
5.98 |
76.353 |
13.255 |
T P |
| 2pm9_B |
280 |
282 |
232 |
1.66 |
6.03 |
75.415 |
13.177 |
T P |
| 3bg1_A |
285 |
282 |
228 |
1.63 |
7.89 |
75.159 |
13.193 |
T P |
| 1nr0_A |
610 |
282 |
232 |
1.96 |
10.34 |
68.604 |
11.254 |
T P |
| 1nex_B |
444 |
282 |
222 |
1.99 |
7.21 |
67.166 |
10.632 |
T P |
| 2ovr_B |
442 |
282 |
216 |
2.01 |
7.41 |
65.704 |
10.235 |
T P |
| 3bws_A |
407 |
282 |
235 |
1.94 |
8.51 |
64.912 |
11.499 |
T P |
| 1pi6_A |
606 |
282 |
235 |
2.07 |
9.36 |
64.606 |
10.807 |
T P |
| 2b4e_A |
386 |
282 |
220 |
2.06 |
10.45 |
62.124 |
10.181 |
T P |
| 2aq5_A |
395 |
282 |
219 |
2.05 |
10.50 |
62.023 |
10.189 |
T P |
| 2zkq_a |
306 |
282 |
226 |
2.44 |
12.83 |
57.615 |
8.899 |
T P |
| 1r5m_A |
351 |
282 |
197 |
2.22 |
8.12 |
53.461 |
8.475 |
T P |
| 1npe_A |
263 |
282 |
182 |
2.26 |
5.49 |
47.581 |
7.723 |
T P |
| 1ijq_A |
308 |
282 |
181 |
2.15 |
8.29 |
45.559 |
8.060 |
T P |
| 1q7f_B |
282 |
282 |
181 |
2.27 |
11.60 |
44.894 |
7.631 |
T P |
| 1rwi_A |
256 |
282 |
174 |
2.36 |
9.77 |
43.893 |
7.081 |
T P |
| 1n7d_A |
639 |
282 |
181 |
2.23 |
6.08 |
43.614 |
7.785 |
T P |
| 1r5x_A |
103 |
282 |
38 |
2.41 |
7.89 |
9.714 |
1.513 |
T P |
| 1oi0_A |
108 |
282 |
31 |
2.71 |
6.45 |
7.091 |
1.102 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]