LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_475.5wLII_11346_58
Total number of 3D structures: 52
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
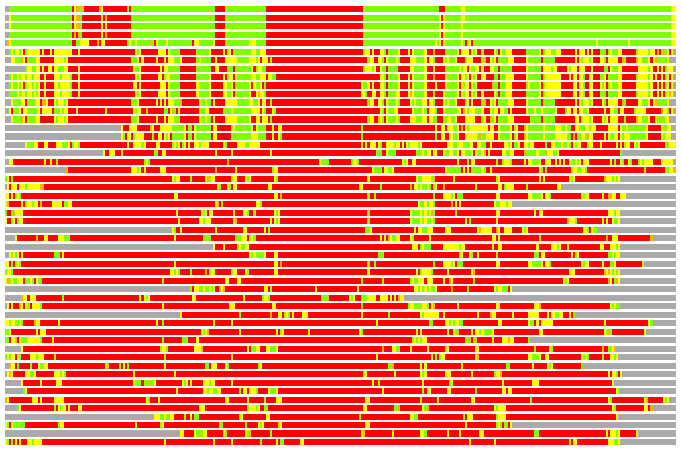
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3fcs_A |
913 |
329 |
250 |
0.96 |
16.80 |
75.540 |
23.689 |
T P |
| 3fcu_A |
457 |
329 |
250 |
0.93 |
16.80 |
75.383 |
24.163 |
T P |
| 1txv_A |
452 |
329 |
250 |
0.92 |
16.40 |
75.244 |
24.395 |
T P |
| 2vdr_A |
452 |
329 |
249 |
0.87 |
16.87 |
75.208 |
25.715 |
T P |
| 1jv2_A |
927 |
329 |
252 |
1.32 |
13.10 |
73.192 |
17.751 |
T P |
| 2w8b_B |
409 |
329 |
157 |
2.35 |
6.37 |
31.361 |
6.414 |
T P |
| 2qr5_A |
576 |
329 |
157 |
2.30 |
9.55 |
30.972 |
6.538 |
T P |
| 2w8b_A |
409 |
329 |
151 |
2.25 |
6.62 |
30.952 |
6.424 |
T P |
| 2hqs_A |
412 |
329 |
159 |
2.32 |
5.66 |
30.849 |
6.581 |
T P |
| 1crz_A |
403 |
329 |
154 |
2.36 |
5.84 |
30.474 |
6.272 |
T P |
| 1c5k_A |
397 |
329 |
159 |
2.34 |
5.66 |
30.454 |
6.516 |
T P |
| 2hu5_B |
575 |
329 |
155 |
2.31 |
9.68 |
30.400 |
6.428 |
T P |
| 2qzp_B |
561 |
329 |
155 |
2.32 |
5.81 |
30.270 |
6.392 |
T P |
| 2hu8_A |
575 |
329 |
149 |
2.35 |
9.40 |
29.634 |
6.089 |
T P |
| 2zux_A |
582 |
329 |
141 |
2.26 |
9.93 |
29.551 |
5.967 |
T P |
| 2zuy_A |
604 |
329 |
143 |
2.25 |
11.19 |
29.467 |
6.076 |
T P |
| 2bwr_A |
401 |
329 |
140 |
2.58 |
5.71 |
26.405 |
5.228 |
T P |
| 1yo8_A |
621 |
329 |
73 |
2.42 |
4.11 |
15.780 |
2.893 |
T P |
| 2rhp_A |
622 |
329 |
74 |
2.54 |
6.76 |
14.366 |
2.804 |
T P |
| 1m1j_B |
402 |
329 |
60 |
2.28 |
8.33 |
12.682 |
2.526 |
T P |
| 1lfa_A |
183 |
329 |
47 |
2.65 |
4.26 |
9.630 |
1.706 |
T P |
| 1pt6_B |
195 |
329 |
50 |
2.74 |
8.00 |
9.441 |
1.759 |
T P |
| 1na5_A |
195 |
329 |
47 |
2.71 |
4.26 |
9.282 |
1.671 |
T P |
| 1qc5_B |
190 |
329 |
45 |
2.67 |
2.22 |
9.122 |
1.622 |
T P |
| 1mf7_A |
194 |
329 |
46 |
2.62 |
13.04 |
9.075 |
1.691 |
T P |
| 2ica_A |
179 |
329 |
46 |
2.67 |
6.52 |
9.012 |
1.661 |
T P |
| 3c96_A |
381 |
329 |
44 |
2.75 |
9.09 |
8.998 |
1.544 |
T P |
| 2b2x_A |
188 |
329 |
44 |
2.66 |
6.82 |
8.995 |
1.593 |
T P |
| 1ido_A |
184 |
329 |
44 |
2.60 |
4.55 |
8.938 |
1.627 |
T P |
| 2qds_A |
224 |
329 |
42 |
2.38 |
14.29 |
8.915 |
1.692 |
T P |
| 1jlm_A |
187 |
329 |
43 |
2.49 |
9.30 |
8.863 |
1.661 |
T P |
| 3bqn_B |
182 |
329 |
43 |
2.73 |
4.65 |
8.795 |
1.521 |
T P |
| 1aox_A |
201 |
329 |
45 |
2.70 |
2.22 |
8.755 |
1.608 |
T P |
| 1qc5_A |
192 |
329 |
40 |
2.43 |
10.00 |
8.624 |
1.584 |
T P |
| 1n3y_A |
189 |
329 |
41 |
2.74 |
4.88 |
8.524 |
1.445 |
T P |
| 1dzi_A |
185 |
329 |
41 |
2.54 |
14.63 |
8.478 |
1.554 |
T P |
| 1mq9_A |
173 |
329 |
43 |
2.82 |
6.98 |
8.418 |
1.472 |
T P |
| 1mq8_B |
177 |
329 |
44 |
2.76 |
4.55 |
8.270 |
1.540 |
T P |
| 3e2m_A |
182 |
329 |
41 |
2.83 |
9.76 |
8.118 |
1.400 |
T P |
| 1ck4_B |
194 |
329 |
42 |
2.70 |
7.14 |
8.093 |
1.501 |
T P |
| 1m1u_A |
184 |
329 |
39 |
2.66 |
2.56 |
8.007 |
1.412 |
T P |
| 1qcy_A |
193 |
329 |
42 |
2.71 |
0.00 |
7.988 |
1.494 |
T P |
| 3bn3_A |
180 |
329 |
38 |
2.53 |
7.89 |
7.855 |
1.448 |
T P |
| 1bho_1 |
189 |
329 |
35 |
2.80 |
0.00 |
7.570 |
1.207 |
T P |
| 1v7p_C |
193 |
329 |
36 |
2.70 |
8.33 |
7.496 |
1.284 |
T P |
| 1n9z_A |
184 |
329 |
37 |
2.60 |
0.00 |
7.443 |
1.372 |
T P |
| 2g23_A |
587 |
329 |
35 |
2.82 |
8.57 |
7.425 |
1.201 |
T P |
| 1t0p_A |
174 |
329 |
40 |
2.84 |
7.50 |
7.370 |
1.361 |
T P |
| 1mjn_A |
179 |
329 |
33 |
2.57 |
0.00 |
7.114 |
1.238 |
T P |
| 1xuo_A |
181 |
329 |
32 |
2.37 |
9.38 |
6.897 |
1.298 |
T P |
| 1rd4_A |
184 |
329 |
33 |
2.77 |
3.03 |
6.550 |
1.152 |
T P |
| 1mhp_B |
192 |
329 |
32 |
2.69 |
6.25 |
6.534 |
1.148 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]