LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_476.5wLII_11346_83
Total number of 3D structures: 81
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
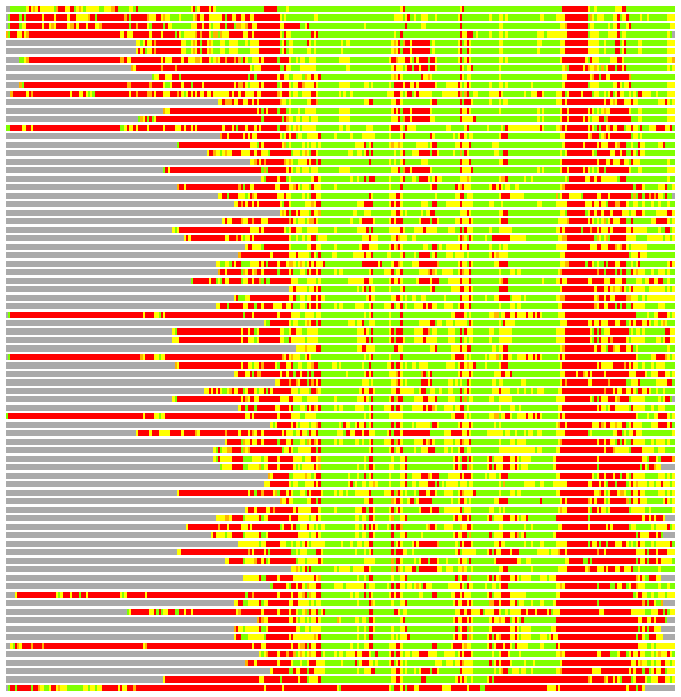
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qzz_A |
340 |
293 |
255 |
1.57 |
20.78 |
83.456 |
15.282 |
T P |
| 1tw3_A |
340 |
293 |
209 |
1.96 |
18.18 |
60.195 |
10.128 |
T P |
| 1xds_A |
336 |
293 |
201 |
1.94 |
19.90 |
59.830 |
9.861 |
T P |
| 2r3s_A |
330 |
293 |
189 |
2.07 |
16.40 |
53.370 |
8.703 |
T P |
| 1fpx_A |
345 |
293 |
183 |
1.99 |
16.39 |
51.701 |
8.750 |
T P |
| 1fp2_A |
345 |
293 |
182 |
1.95 |
16.48 |
51.636 |
8.892 |
T P |
| 1kyz_E |
361 |
293 |
179 |
2.16 |
13.97 |
50.995 |
7.922 |
T P |
| 1fpq_A |
333 |
293 |
156 |
1.82 |
19.87 |
48.932 |
8.116 |
T P |
| 2ip2_A |
330 |
293 |
154 |
1.83 |
18.18 |
47.464 |
7.987 |
T P |
| 1fp1_D |
340 |
293 |
162 |
2.18 |
18.52 |
46.930 |
7.118 |
T P |
| 3dp7_A |
351 |
293 |
187 |
2.44 |
11.76 |
45.359 |
7.354 |
T P |
| 1wzn_A |
244 |
293 |
148 |
2.03 |
25.00 |
43.414 |
6.949 |
T P |
| 1zg3_A |
358 |
293 |
143 |
1.88 |
23.08 |
43.284 |
7.222 |
T P |
| 1zgj_A |
354 |
293 |
144 |
1.83 |
22.92 |
43.141 |
7.473 |
T P |
| 1x19_A |
350 |
293 |
163 |
2.36 |
15.95 |
43.012 |
6.638 |
T P |
| 3dh0_B |
190 |
293 |
145 |
1.94 |
18.62 |
42.929 |
7.110 |
T P |
| 1ve3_B |
226 |
293 |
150 |
1.97 |
20.00 |
42.631 |
7.256 |
T P |
| 3e8s_A |
220 |
293 |
148 |
1.97 |
22.30 |
42.506 |
7.139 |
T P |
| 3bus_A |
251 |
293 |
144 |
1.87 |
22.92 |
41.318 |
7.315 |
T P |
| 1zga_A |
357 |
293 |
140 |
1.81 |
23.57 |
41.097 |
7.330 |
T P |
| 2p7i_B |
229 |
293 |
146 |
1.90 |
12.33 |
40.998 |
7.302 |
T P |
| 2pjd_A |
334 |
293 |
133 |
1.92 |
18.05 |
40.469 |
6.572 |
T P |
| 3dlc_A |
219 |
293 |
143 |
2.05 |
17.48 |
40.202 |
6.640 |
T P |
| 3d2l_C |
242 |
293 |
149 |
2.08 |
23.49 |
38.961 |
6.847 |
T P |
| 2gh1_A |
281 |
293 |
137 |
1.99 |
21.17 |
38.798 |
6.550 |
T P |
| 2gs9_A |
211 |
293 |
138 |
2.09 |
15.22 |
38.186 |
6.307 |
T P |
| 1d2g_A |
292 |
293 |
147 |
2.18 |
10.88 |
38.181 |
6.459 |
T P |
| 3f4k_A |
254 |
293 |
146 |
2.27 |
18.49 |
38.080 |
6.163 |
T P |
| 1y8c_A |
246 |
293 |
147 |
2.20 |
17.69 |
37.940 |
6.404 |
T P |
| 3cgg_A |
186 |
293 |
129 |
2.00 |
18.60 |
37.554 |
6.131 |
T P |
| 1vlm_A |
207 |
293 |
144 |
2.14 |
14.58 |
37.273 |
6.442 |
T P |
| 1tpy_A |
285 |
293 |
142 |
2.15 |
16.90 |
37.250 |
6.313 |
T P |
| 2avn_A |
247 |
293 |
144 |
2.12 |
18.06 |
37.225 |
6.478 |
T P |
| 1xxl_A |
234 |
293 |
135 |
1.98 |
18.52 |
36.972 |
6.480 |
T P |
| 3e7p_A |
253 |
293 |
139 |
2.20 |
19.42 |
36.959 |
6.044 |
T P |
| 1kph_B |
285 |
293 |
144 |
2.17 |
18.75 |
36.867 |
6.353 |
T P |
| 1nv8_A |
271 |
293 |
130 |
2.04 |
18.46 |
36.697 |
6.087 |
T P |
| 1ri5_A |
252 |
293 |
141 |
2.17 |
17.02 |
36.647 |
6.215 |
T P |
| 1r74_B |
279 |
293 |
144 |
2.01 |
11.11 |
36.530 |
6.827 |
T P |
| 1r8x_B |
284 |
293 |
151 |
2.16 |
11.26 |
36.390 |
6.685 |
T P |
| 1vl5_A |
231 |
293 |
133 |
1.97 |
17.29 |
36.357 |
6.428 |
T P |
| 1sg9_A |
274 |
293 |
131 |
2.06 |
16.79 |
36.328 |
6.069 |
T P |
| 2o57_A |
282 |
293 |
145 |
2.13 |
18.62 |
36.287 |
6.488 |
T P |
| 2yqz_A |
261 |
293 |
128 |
2.09 |
15.62 |
35.799 |
5.838 |
T P |
| 3dtn_A |
220 |
293 |
130 |
2.06 |
16.92 |
35.455 |
6.008 |
T P |
| 2ex4_A |
221 |
293 |
147 |
2.24 |
14.97 |
35.433 |
6.287 |
T P |
| 1xva_A |
292 |
293 |
146 |
2.11 |
10.27 |
35.342 |
6.611 |
T P |
| 1l1e_A |
272 |
293 |
142 |
2.17 |
19.72 |
35.339 |
6.256 |
T P |
| 1vq1_A |
266 |
293 |
133 |
2.15 |
16.54 |
35.320 |
5.905 |
T P |
| 2p8j_B |
207 |
293 |
134 |
2.14 |
17.16 |
35.170 |
5.981 |
T P |
| 2qyo_A |
353 |
293 |
142 |
2.30 |
15.49 |
35.059 |
5.926 |
T P |
| 1kpg_A |
285 |
293 |
147 |
2.27 |
19.05 |
34.956 |
6.211 |
T P |
| 1kpi_A |
291 |
293 |
144 |
2.31 |
15.97 |
34.793 |
5.970 |
T P |
| 1f3l_A |
321 |
293 |
127 |
2.04 |
21.26 |
34.789 |
5.928 |
T P |
| 2fyt_A |
311 |
293 |
126 |
2.04 |
22.22 |
34.783 |
5.899 |
T P |
| 3ccf_B |
242 |
293 |
129 |
2.02 |
22.48 |
34.754 |
6.089 |
T P |
| 3bgv_B |
271 |
293 |
142 |
2.28 |
14.79 |
34.631 |
5.958 |
T P |
| 2fk8_A |
281 |
293 |
140 |
2.23 |
15.00 |
34.570 |
6.021 |
T P |
| 3ege_A |
247 |
293 |
126 |
1.94 |
17.46 |
34.486 |
6.186 |
T P |
| 3bxo_A |
236 |
293 |
134 |
2.17 |
18.66 |
34.294 |
5.916 |
T P |
| 2v74_D |
332 |
293 |
122 |
2.07 |
18.85 |
34.275 |
5.629 |
T P |
| 1im8_A |
225 |
293 |
138 |
2.13 |
21.01 |
33.905 |
6.188 |
T P |
| 3b3f_A |
337 |
293 |
124 |
2.02 |
17.74 |
33.869 |
5.858 |
T P |
| 3dli_A |
221 |
293 |
146 |
2.25 |
15.07 |
33.636 |
6.210 |
T P |
| 1nkv_C |
249 |
293 |
119 |
2.12 |
21.01 |
33.123 |
5.349 |
T P |
| 1p91_A |
268 |
293 |
125 |
2.12 |
16.00 |
32.951 |
5.643 |
T P |
| 3cc8_A |
211 |
293 |
133 |
2.15 |
19.55 |
32.605 |
5.918 |
T P |
| 1or8_A |
313 |
293 |
118 |
2.11 |
18.64 |
32.520 |
5.343 |
T P |
| 3bkw_A |
219 |
293 |
131 |
2.21 |
23.66 |
32.478 |
5.667 |
T P |
| 3cjt_A |
254 |
293 |
128 |
2.21 |
21.88 |
31.862 |
5.541 |
T P |
| 1g6q_1 |
328 |
293 |
121 |
2.04 |
18.18 |
31.765 |
5.646 |
T P |
| 2as0_A |
396 |
293 |
127 |
2.25 |
18.11 |
31.378 |
5.397 |
T P |
| 3b3j_A |
332 |
293 |
113 |
1.98 |
19.47 |
31.298 |
5.431 |
T P |
| 1orh_A |
318 |
293 |
118 |
2.04 |
16.10 |
31.240 |
5.517 |
T P |
| 1ori_A |
316 |
293 |
121 |
2.13 |
17.36 |
30.641 |
5.428 |
T P |
| 2nxc_A |
249 |
293 |
121 |
2.24 |
25.62 |
30.541 |
5.167 |
T P |
| 3g5l_A |
222 |
293 |
128 |
2.28 |
19.53 |
30.514 |
5.367 |
T P |
| 1l3i_A |
186 |
293 |
122 |
2.22 |
20.49 |
30.129 |
5.262 |
T P |
| 2p35_A |
245 |
293 |
117 |
2.19 |
23.93 |
29.134 |
5.116 |
T P |
| 1m6y_A |
293 |
293 |
98 |
1.95 |
21.43 |
28.570 |
4.772 |
T P |
| 1jmr_B |
247 |
293 |
58 |
2.57 |
10.34 |
14.415 |
2.175 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]