LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_478.5wLII_11346_90
Total number of 3D structures: 54
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
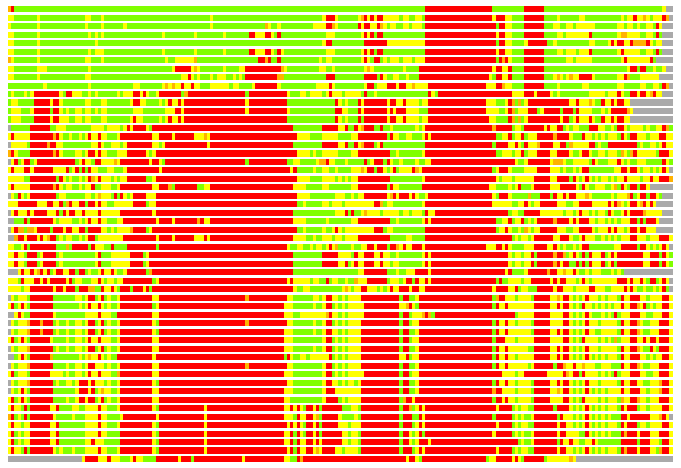
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2otd_A |
245 |
207 |
177 |
0.53 |
20.34 |
85.094 |
27.896 |
T P |
| 2pz0_B |
243 |
207 |
167 |
1.61 |
16.77 |
76.612 |
9.787 |
T P |
| 2oog_D |
268 |
207 |
167 |
1.74 |
14.97 |
73.593 |
9.075 |
T P |
| 1ydy_A |
328 |
207 |
163 |
1.78 |
11.66 |
72.195 |
8.693 |
T P |
| 1zcc_A |
240 |
207 |
160 |
1.68 |
11.88 |
72.027 |
8.988 |
T P |
| 1t8q_B |
329 |
207 |
162 |
1.75 |
11.73 |
71.928 |
8.734 |
T P |
| 3ch0_A |
272 |
207 |
163 |
1.84 |
10.43 |
70.885 |
8.387 |
T P |
| 1vd6_A |
218 |
207 |
153 |
1.62 |
19.61 |
68.294 |
8.882 |
T P |
| 1o1z_A |
226 |
207 |
148 |
1.80 |
13.51 |
65.419 |
7.797 |
T P |
| 2o55_A |
254 |
207 |
162 |
2.01 |
14.20 |
60.479 |
7.684 |
T P |
| 1xx1_A |
285 |
207 |
115 |
2.32 |
11.30 |
36.702 |
4.747 |
T P |
| 1djx_B |
561 |
207 |
103 |
2.16 |
8.74 |
36.671 |
4.551 |
T P |
| 2zkm_X |
708 |
207 |
105 |
2.23 |
6.67 |
36.567 |
4.497 |
T P |
| 1qas_A |
505 |
207 |
102 |
2.14 |
10.78 |
36.171 |
4.545 |
T P |
| 1xrt_A |
369 |
207 |
99 |
2.18 |
8.08 |
33.106 |
4.349 |
T P |
| 1k1d_A |
459 |
207 |
101 |
2.48 |
4.95 |
31.343 |
3.912 |
T P |
| 3d6n_A |
422 |
207 |
95 |
2.30 |
9.47 |
31.307 |
3.953 |
T P |
| 1yny_B |
459 |
207 |
98 |
2.39 |
7.14 |
31.037 |
3.940 |
T P |
| 1v4y_A |
474 |
207 |
98 |
2.41 |
10.20 |
30.622 |
3.899 |
T P |
| 1ybq_A |
389 |
207 |
100 |
2.42 |
4.00 |
30.586 |
3.974 |
T P |
| 3e74_B |
432 |
207 |
98 |
2.44 |
6.12 |
30.400 |
3.855 |
T P |
| 2fty_A |
531 |
207 |
96 |
2.43 |
3.12 |
30.282 |
3.791 |
T P |
| 2qt3_A |
401 |
207 |
97 |
2.33 |
11.34 |
30.060 |
3.992 |
T P |
| 1m7j_A |
474 |
207 |
100 |
2.58 |
5.00 |
30.053 |
3.730 |
T P |
| 2g3f_A |
414 |
207 |
100 |
2.72 |
12.00 |
30.051 |
3.551 |
T P |
| 1gkp_A |
457 |
207 |
91 |
2.36 |
7.69 |
29.823 |
3.701 |
T P |
| 1rk6_A |
474 |
207 |
94 |
2.55 |
5.32 |
29.689 |
3.553 |
T P |
| 1nfg_A |
456 |
207 |
96 |
2.51 |
8.33 |
29.386 |
3.678 |
T P |
| 1ybq_B |
388 |
207 |
92 |
2.27 |
6.52 |
29.199 |
3.890 |
T P |
| 1a5l_C |
536 |
207 |
89 |
2.34 |
6.74 |
29.176 |
3.648 |
T P |
| 1ejx_C |
555 |
207 |
87 |
2.28 |
6.90 |
28.936 |
3.662 |
T P |
| 2ogj_A |
378 |
207 |
83 |
2.35 |
8.43 |
28.631 |
3.388 |
T P |
| 1rjq_A |
474 |
207 |
95 |
2.64 |
11.58 |
28.356 |
3.470 |
T P |
| 1onw_B |
379 |
207 |
92 |
2.55 |
5.43 |
28.096 |
3.475 |
T P |
| 1a5k_C |
566 |
207 |
92 |
2.47 |
9.78 |
27.636 |
3.581 |
T P |
| 1e9y_B |
568 |
207 |
93 |
2.55 |
3.23 |
27.383 |
3.505 |
T P |
| 1a5m_C |
566 |
207 |
92 |
2.54 |
8.70 |
27.268 |
3.486 |
T P |
| 1ejt_C |
565 |
207 |
93 |
2.58 |
9.68 |
27.204 |
3.467 |
T P |
| 1ejr_C |
552 |
207 |
92 |
2.55 |
9.78 |
27.093 |
3.468 |
T P |
| 1fwf_C |
550 |
207 |
90 |
2.55 |
8.89 |
27.046 |
3.393 |
T P |
| 1krb_C |
565 |
207 |
90 |
2.50 |
10.00 |
27.012 |
3.459 |
T P |
| 4ubp_C |
569 |
207 |
91 |
2.58 |
5.49 |
26.928 |
3.401 |
T P |
| 1eju_C |
552 |
207 |
92 |
2.62 |
7.61 |
26.770 |
3.378 |
T P |
| 1krc_C |
566 |
207 |
90 |
2.58 |
8.89 |
26.561 |
3.357 |
T P |
| 1ejs_C |
565 |
207 |
90 |
2.56 |
7.78 |
26.495 |
3.379 |
T P |
| 2kau_C |
565 |
207 |
89 |
2.50 |
8.99 |
26.376 |
3.420 |
T P |
| 1ie7_C |
569 |
207 |
86 |
2.46 |
6.98 |
25.574 |
3.362 |
T P |
| 1fwh_C |
565 |
207 |
80 |
2.38 |
8.75 |
25.285 |
3.231 |
T P |
| 1e9z_B |
568 |
207 |
80 |
2.41 |
6.25 |
25.258 |
3.192 |
T P |
| 1fwa_C |
565 |
207 |
81 |
2.44 |
8.64 |
25.158 |
3.192 |
T P |
| 1fwg_C |
565 |
207 |
78 |
2.42 |
8.97 |
25.069 |
3.100 |
T P |
| 1ejv_C |
552 |
207 |
75 |
2.40 |
13.33 |
24.368 |
3.005 |
T P |
| 1ef2_A |
565 |
207 |
79 |
2.45 |
8.86 |
24.331 |
3.102 |
T P |
| 2fjl_A |
150 |
207 |
44 |
2.59 |
11.36 |
14.460 |
1.636 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]