LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_489.5wLII_11346_157
Total number of 3D structures: 40
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
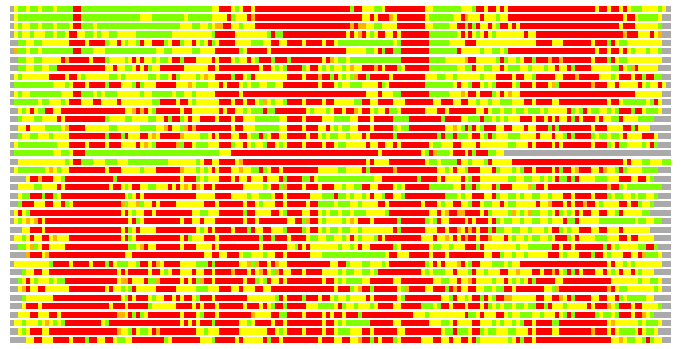
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2ibd_A |
192 |
167 |
90 |
2.07 |
10.00 |
43.859 |
4.157 |
T P |
| 2eh3_A |
171 |
167 |
89 |
2.13 |
12.36 |
41.450 |
3.985 |
T P |
| 3ccy_A |
192 |
167 |
92 |
2.27 |
15.22 |
41.274 |
3.888 |
T P |
| 1pb6_A |
198 |
167 |
98 |
2.51 |
9.18 |
41.032 |
3.751 |
T P |
| 3bcg_A |
207 |
167 |
105 |
2.55 |
10.48 |
40.910 |
3.967 |
T P |
| 2iai_A |
201 |
167 |
94 |
2.18 |
10.64 |
40.365 |
4.131 |
T P |
| 2uxu_A |
206 |
167 |
99 |
2.46 |
14.14 |
39.616 |
3.874 |
T P |
| 1vi0_A |
189 |
167 |
98 |
2.56 |
12.24 |
38.990 |
3.691 |
T P |
| 2fbq_A |
213 |
167 |
99 |
2.60 |
9.09 |
38.557 |
3.671 |
T P |
| 2id6_A |
202 |
167 |
96 |
2.47 |
10.42 |
37.637 |
3.738 |
T P |
| 3cdl_B |
188 |
167 |
82 |
2.39 |
14.63 |
37.448 |
3.291 |
T P |
| 3dew_A |
192 |
167 |
96 |
2.54 |
10.42 |
36.767 |
3.631 |
T P |
| 2g3b_A |
188 |
167 |
92 |
2.62 |
14.13 |
36.372 |
3.380 |
T P |
| 2ofl_A |
221 |
167 |
94 |
2.51 |
5.32 |
35.993 |
3.599 |
T P |
| 3c07_A |
221 |
167 |
91 |
2.72 |
5.49 |
35.831 |
3.230 |
T P |
| 3bt9_B |
186 |
167 |
93 |
2.61 |
7.53 |
35.462 |
3.426 |
T P |
| 3f1b_A |
183 |
167 |
96 |
2.58 |
9.38 |
35.300 |
3.579 |
T P |
| 2qwt_A |
167 |
167 |
66 |
1.76 |
10.61 |
35.180 |
3.550 |
T P |
| 3geu_A |
189 |
167 |
85 |
2.61 |
9.41 |
34.584 |
3.138 |
T P |
| 3br5_E |
186 |
167 |
93 |
2.68 |
5.38 |
34.480 |
3.351 |
T P |
| 3bru_A |
191 |
167 |
86 |
2.44 |
5.81 |
34.406 |
3.388 |
T P |
| 2gen_A |
188 |
167 |
86 |
2.65 |
8.14 |
33.767 |
3.132 |
T P |
| 3eup_A |
203 |
167 |
88 |
2.48 |
9.09 |
33.536 |
3.405 |
T P |
| 1sgm_A |
184 |
167 |
87 |
2.70 |
5.75 |
33.444 |
3.109 |
T P |
| 3br3_A |
185 |
167 |
84 |
2.62 |
10.71 |
33.361 |
3.090 |
T P |
| 3bhq_A |
202 |
167 |
82 |
2.58 |
7.32 |
33.281 |
3.065 |
T P |
| 3br0_A |
186 |
167 |
84 |
2.64 |
9.52 |
33.121 |
3.070 |
T P |
| 2g7s_A |
190 |
167 |
84 |
2.78 |
8.33 |
31.825 |
2.921 |
T P |
| 2iek_A |
199 |
167 |
83 |
2.70 |
13.25 |
31.821 |
2.964 |
T P |
| 1zkg_A |
200 |
167 |
76 |
2.50 |
7.89 |
31.779 |
2.918 |
T P |
| 3g7r_B |
202 |
167 |
85 |
2.76 |
7.06 |
31.214 |
2.969 |
T P |
| 1rkt_A |
204 |
167 |
83 |
2.80 |
8.43 |
30.883 |
2.858 |
T P |
| 2zcn_D |
184 |
167 |
81 |
2.74 |
9.88 |
29.902 |
2.850 |
T P |
| 2qib_A |
222 |
167 |
78 |
2.92 |
12.82 |
29.633 |
2.583 |
T P |
| 2f07_A |
197 |
167 |
79 |
2.84 |
15.19 |
29.541 |
2.690 |
T P |
| 3dpj_A |
194 |
167 |
78 |
2.72 |
6.41 |
29.490 |
2.771 |
T P |
| 2hyt_A |
193 |
167 |
74 |
2.66 |
5.41 |
29.359 |
2.677 |
T P |
| 2zcm_B |
180 |
167 |
75 |
2.51 |
10.67 |
29.131 |
2.878 |
T P |
| 3dcf_A |
185 |
167 |
76 |
2.67 |
6.58 |
27.969 |
2.748 |
T P |
| 2i10_B |
182 |
167 |
70 |
2.88 |
5.71 |
26.468 |
2.346 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]