LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_50.5wLII_10933_46
Total number of 3D structures: 46
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
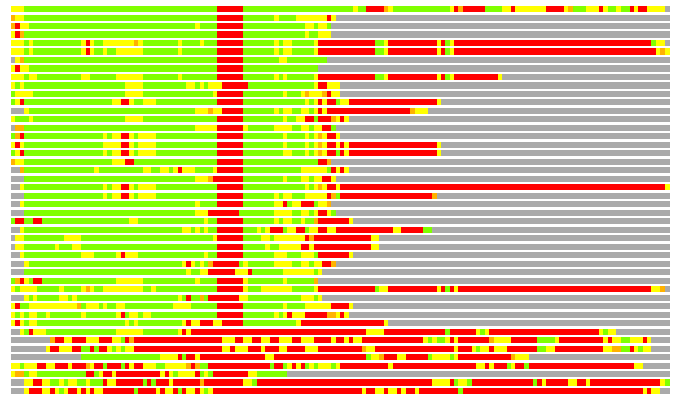
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1y9q_A |
178 |
150 |
124 |
1.63 |
19.35 |
73.602 |
7.181 |
T P |
| 3clc_B |
77 |
150 |
67 |
1.54 |
10.45 |
42.746 |
4.077 |
T P |
| 2b5a_A |
77 |
150 |
66 |
1.29 |
18.18 |
42.626 |
4.754 |
T P |
| 3f52_E |
78 |
150 |
66 |
1.37 |
22.73 |
42.094 |
4.497 |
T P |
| 1lli_B |
92 |
150 |
72 |
1.99 |
15.28 |
41.599 |
3.439 |
T P |
| 1rio_A |
97 |
150 |
72 |
1.99 |
15.28 |
41.216 |
3.444 |
T P |
| 1adr_A |
76 |
150 |
65 |
1.31 |
7.69 |
41.196 |
4.608 |
T P |
| 1y7y_A |
69 |
150 |
63 |
0.96 |
20.63 |
41.187 |
5.920 |
T P |
| 1lmb_4 |
92 |
150 |
71 |
1.91 |
16.90 |
41.125 |
3.539 |
T P |
| 2bnm_A |
194 |
150 |
67 |
1.74 |
16.42 |
40.959 |
3.639 |
T P |
| 3f6w_A |
82 |
150 |
68 |
1.94 |
11.76 |
40.562 |
3.333 |
T P |
| 2axz_A |
306 |
150 |
66 |
1.74 |
13.64 |
40.443 |
3.584 |
T P |
| 1b0n_A |
103 |
150 |
67 |
1.79 |
17.91 |
40.321 |
3.549 |
T P |
| 3eus_A |
85 |
150 |
65 |
1.72 |
10.77 |
40.096 |
3.570 |
T P |
| 2r1j_L |
66 |
150 |
65 |
1.73 |
7.69 |
40.058 |
3.554 |
T P |
| 2axu_A |
303 |
150 |
64 |
1.73 |
14.06 |
39.960 |
3.501 |
T P |
| 2grm_A |
315 |
150 |
66 |
1.81 |
13.64 |
39.803 |
3.447 |
T P |
| 2aw6_A |
287 |
150 |
66 |
1.82 |
13.64 |
39.761 |
3.442 |
T P |
| 2ewt_A |
71 |
150 |
63 |
1.63 |
17.46 |
39.690 |
3.633 |
T P |
| 3b7h_A |
76 |
150 |
66 |
1.97 |
12.12 |
39.331 |
3.189 |
T P |
| 2cro_A |
65 |
150 |
62 |
1.56 |
16.13 |
39.307 |
3.740 |
T P |
| 2awi_A |
298 |
150 |
64 |
1.79 |
14.06 |
38.938 |
3.384 |
T P |
| 2axv_B |
303 |
150 |
64 |
1.89 |
14.06 |
38.926 |
3.222 |
T P |
| 1x57_A |
91 |
150 |
61 |
1.56 |
14.75 |
38.749 |
3.685 |
T P |
| 1r69_A |
63 |
150 |
62 |
1.55 |
19.35 |
38.749 |
3.756 |
T P |
| 1utx_A |
66 |
150 |
61 |
1.52 |
16.39 |
38.538 |
3.760 |
T P |
| 2jvl_A |
107 |
150 |
63 |
1.79 |
7.94 |
38.490 |
3.328 |
T P |
| 3cec_A |
91 |
150 |
63 |
1.84 |
7.94 |
38.489 |
3.253 |
T P |
| 2eby_A |
102 |
150 |
62 |
1.79 |
14.52 |
38.444 |
3.279 |
T P |
| 2qfc_A |
284 |
150 |
62 |
1.72 |
14.52 |
37.994 |
3.413 |
T P |
| 1r63_A |
63 |
150 |
62 |
1.73 |
20.97 |
37.988 |
3.382 |
T P |
| 1sq8_A |
64 |
150 |
61 |
1.61 |
21.31 |
37.942 |
3.560 |
T P |
| 3bs3_A |
62 |
150 |
61 |
1.70 |
6.56 |
37.275 |
3.387 |
T P |
| 3bdn_A |
234 |
150 |
73 |
2.27 |
16.44 |
36.577 |
3.082 |
T P |
| 2r63_A |
63 |
150 |
60 |
1.73 |
18.33 |
36.442 |
3.273 |
T P |
| 2ofy_A |
82 |
150 |
67 |
2.20 |
17.91 |
36.379 |
2.913 |
T P |
| 2ef8_A |
84 |
150 |
63 |
2.02 |
14.29 |
35.942 |
2.966 |
T P |
| 3dnv_B |
71 |
150 |
57 |
1.72 |
10.53 |
35.222 |
3.126 |
T P |
| 2k9q_A |
77 |
150 |
49 |
2.17 |
18.37 |
25.817 |
2.160 |
T P |
| 1scj_A |
275 |
150 |
53 |
2.82 |
9.43 |
22.560 |
1.818 |
T P |
| 1mee_A |
275 |
150 |
49 |
2.81 |
4.08 |
22.009 |
1.684 |
T P |
| 2zhg_A |
121 |
150 |
43 |
2.25 |
13.95 |
21.594 |
1.832 |
T P |
| 2h5g_B |
425 |
150 |
49 |
2.81 |
14.29 |
21.525 |
1.683 |
T P |
| 2p5t_E |
95 |
150 |
39 |
2.31 |
12.82 |
18.846 |
1.616 |
T P |
| 2ras_B |
204 |
150 |
40 |
2.50 |
10.00 |
17.907 |
1.537 |
T P |
| 1nr3_A |
122 |
150 |
31 |
3.03 |
6.45 |
14.262 |
0.990 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]