LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_501.5wLII_11364_25
Total number of 3D structures: 36
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
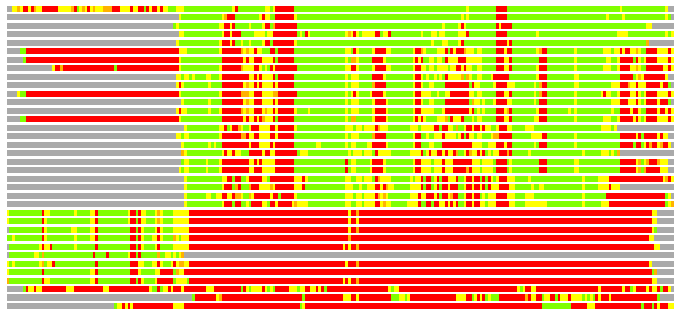
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ksk_A |
234 |
248 |
211 |
1.81 |
21.33 |
81.090 |
11.024 |
T P |
| 1vio_A |
230 |
248 |
168 |
1.02 |
22.02 |
66.377 |
15.017 |
T P |
| 2oml_A |
181 |
248 |
159 |
1.30 |
33.96 |
61.053 |
11.340 |
T P |
| 2gml_A |
175 |
248 |
160 |
1.65 |
30.00 |
59.506 |
9.121 |
T P |
| 2olw_A |
179 |
248 |
156 |
1.38 |
34.62 |
59.467 |
10.565 |
T P |
| 2hvy_A |
320 |
248 |
143 |
1.97 |
20.98 |
46.688 |
6.915 |
T P |
| 2aus_C |
311 |
248 |
138 |
1.90 |
20.29 |
45.812 |
6.906 |
T P |
| 2rfk_A |
334 |
248 |
140 |
1.98 |
21.43 |
45.423 |
6.742 |
T P |
| 1r3e_A |
305 |
248 |
135 |
1.96 |
18.52 |
45.308 |
6.548 |
T P |
| 1zl3_A |
302 |
248 |
133 |
1.91 |
18.80 |
45.304 |
6.609 |
T P |
| 2apo_A |
304 |
248 |
139 |
2.06 |
20.14 |
44.336 |
6.425 |
T P |
| 1k8w_A |
304 |
248 |
131 |
1.83 |
19.08 |
44.174 |
6.783 |
T P |
| 1r3f_A |
278 |
248 |
131 |
1.97 |
19.08 |
42.997 |
6.315 |
T P |
| 2ey4_A |
329 |
248 |
142 |
1.94 |
20.42 |
42.307 |
6.946 |
T P |
| 1v9k_A |
227 |
248 |
134 |
2.09 |
20.90 |
40.681 |
6.114 |
T P |
| 1ze1_A |
308 |
248 |
136 |
2.01 |
18.38 |
40.674 |
6.431 |
T P |
| 1s71_B |
266 |
248 |
123 |
1.90 |
21.95 |
40.653 |
6.158 |
T P |
| 2i82_A |
217 |
248 |
132 |
2.01 |
18.94 |
40.161 |
6.258 |
T P |
| 2ab4_A |
300 |
248 |
131 |
1.98 |
17.56 |
39.227 |
6.303 |
T P |
| 1ze2_A |
300 |
248 |
130 |
1.96 |
18.46 |
39.156 |
6.303 |
T P |
| 1prz_A |
252 |
248 |
125 |
2.01 |
20.80 |
37.278 |
5.920 |
T P |
| 1qyu_A |
250 |
248 |
124 |
2.08 |
20.16 |
37.094 |
5.677 |
T P |
| 1xpi_A |
229 |
248 |
125 |
2.12 |
21.60 |
36.790 |
5.621 |
T P |
| 1v9f_A |
250 |
248 |
126 |
2.16 |
22.22 |
36.232 |
5.570 |
T P |
| 1fjg_D |
208 |
248 |
67 |
1.91 |
20.90 |
23.989 |
3.334 |
T P |
| 2gy9_D |
204 |
248 |
67 |
1.89 |
16.42 |
23.988 |
3.374 |
T P |
| 1n32_D |
208 |
248 |
65 |
1.81 |
21.54 |
23.812 |
3.396 |
T P |
| 1qd7_C |
159 |
248 |
65 |
1.84 |
20.00 |
23.697 |
3.355 |
T P |
| 2qal_D |
205 |
248 |
65 |
1.93 |
13.85 |
23.573 |
3.198 |
T P |
| 1dm9_B |
107 |
248 |
60 |
1.69 |
16.67 |
22.966 |
3.361 |
T P |
| 1vs5_D |
205 |
248 |
64 |
2.10 |
14.06 |
22.535 |
2.909 |
T P |
| 2vqe_D |
208 |
248 |
58 |
1.56 |
20.69 |
22.071 |
3.488 |
T P |
| 3bbn_D |
199 |
248 |
62 |
1.90 |
12.90 |
21.969 |
3.098 |
T P |
| 1oao_A |
673 |
248 |
60 |
2.75 |
5.00 |
15.103 |
2.102 |
T P |
| 1jyt_A |
163 |
248 |
49 |
2.73 |
10.20 |
13.307 |
1.729 |
T P |
| 1job_A |
162 |
248 |
30 |
2.28 |
6.67 |
9.304 |
1.259 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]