LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_504.5wLII_11364_42
Total number of 3D structures: 42
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
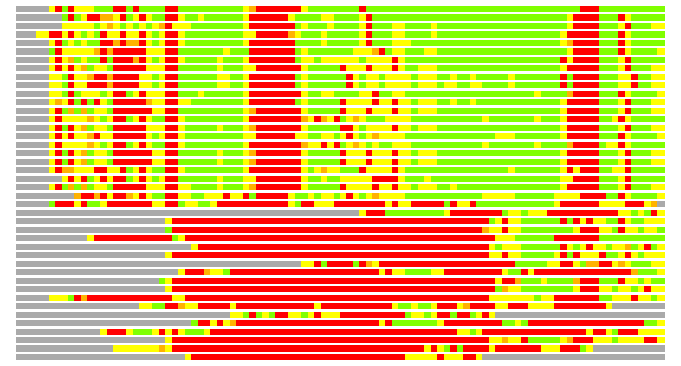
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2vpz_A |
734 |
100 |
77 |
1.35 |
28.57 |
73.842 |
5.325 |
T P |
| 2nya_A |
791 |
100 |
74 |
1.85 |
18.92 |
66.218 |
3.804 |
T P |
| 2e7z_A |
727 |
100 |
79 |
2.12 |
6.33 |
65.835 |
3.558 |
T P |
| 1h0h_A |
976 |
100 |
77 |
1.98 |
15.58 |
65.439 |
3.710 |
T P |
| 1kqf_A |
981 |
100 |
75 |
2.02 |
14.67 |
63.424 |
3.544 |
T P |
| 2ivf_A |
912 |
100 |
73 |
2.06 |
9.59 |
61.847 |
3.381 |
T P |
| 1ti6_A |
875 |
100 |
73 |
2.06 |
9.59 |
60.945 |
3.377 |
T P |
| 1eu1_A |
767 |
100 |
70 |
1.98 |
11.43 |
58.755 |
3.368 |
T P |
| 1y5i_A |
1244 |
100 |
72 |
2.08 |
11.11 |
58.555 |
3.297 |
T P |
| 1q16_A |
1244 |
100 |
71 |
2.03 |
11.27 |
58.489 |
3.336 |
T P |
| 1ogy_A |
789 |
100 |
75 |
2.03 |
14.67 |
58.090 |
3.525 |
T P |
| 1e61_C |
767 |
100 |
69 |
1.99 |
7.25 |
57.847 |
3.307 |
T P |
| 1cxs_A |
768 |
100 |
70 |
2.01 |
11.43 |
55.809 |
3.321 |
T P |
| 2v3v_A |
721 |
100 |
73 |
2.19 |
15.07 |
55.795 |
3.194 |
T P |
| 4dmr_A |
773 |
100 |
70 |
2.04 |
8.57 |
55.202 |
3.267 |
T P |
| 1g8k_A |
822 |
100 |
72 |
2.22 |
15.28 |
55.053 |
3.098 |
T P |
| 2nap_A |
720 |
100 |
74 |
2.21 |
16.22 |
55.028 |
3.208 |
T P |
| 1dmr_A |
779 |
100 |
69 |
2.02 |
7.25 |
54.711 |
3.250 |
T P |
| 1e18_A |
779 |
100 |
69 |
2.03 |
7.25 |
54.707 |
3.243 |
T P |
| 2iv2_X |
696 |
100 |
73 |
2.22 |
8.22 |
54.538 |
3.153 |
T P |
| 1tmo_A |
794 |
100 |
69 |
2.04 |
14.49 |
52.382 |
3.225 |
T P |
| 1dms_A |
766 |
100 |
71 |
2.11 |
7.04 |
52.112 |
3.215 |
T P |
| 1g8j_C |
821 |
100 |
70 |
2.41 |
14.29 |
51.093 |
2.793 |
T P |
| 2fug_3 |
737 |
100 |
43 |
2.35 |
13.95 |
31.912 |
1.754 |
T P |
| 1h95_A |
79 |
100 |
24 |
2.15 |
4.17 |
19.810 |
1.065 |
T P |
| 2i5m_X |
66 |
100 |
23 |
2.12 |
13.04 |
19.072 |
1.038 |
T P |
| 1hz9_A |
66 |
100 |
24 |
2.40 |
16.67 |
18.890 |
0.961 |
T P |
| 1mjc_A |
69 |
100 |
22 |
1.91 |
4.55 |
18.876 |
1.093 |
T P |
| 1mef |
70 |
100 |
25 |
2.45 |
4.00 |
18.875 |
0.982 |
T P |
| 1i5f_A |
66 |
100 |
23 |
2.21 |
8.70 |
18.657 |
0.995 |
T P |
| 2bh8_B |
86 |
100 |
25 |
2.54 |
12.00 |
18.181 |
0.946 |
T P |
| 1hzc_A |
66 |
100 |
23 |
2.52 |
0.00 |
17.977 |
0.876 |
T P |
| 1g6p_A |
66 |
100 |
22 |
2.15 |
18.18 |
17.905 |
0.980 |
T P |
| 1c9o_A |
66 |
100 |
23 |
2.35 |
13.04 |
17.810 |
0.940 |
T P |
| 2ytx_A |
97 |
100 |
26 |
2.84 |
7.69 |
17.748 |
0.885 |
T P |
| 2ytv_A |
79 |
100 |
25 |
2.88 |
8.00 |
17.322 |
0.839 |
T P |
| 3cam_A |
67 |
100 |
22 |
2.38 |
0.00 |
17.205 |
0.888 |
T P |
| 1hza_A |
67 |
100 |
20 |
2.05 |
0.00 |
17.191 |
0.928 |
T P |
| 2i5l_X |
67 |
100 |
23 |
2.55 |
8.70 |
16.285 |
0.867 |
T P |
| 2k5n_A |
74 |
100 |
22 |
2.67 |
18.18 |
16.024 |
0.793 |
T P |
| 2es2_A |
67 |
100 |
19 |
2.80 |
21.05 |
13.142 |
0.654 |
T P |
| 1hzb_A |
66 |
100 |
10 |
2.99 |
20.00 |
7.280 |
0.323 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]